INFORMACION SOBRE LA PROPIEDAD INTELECTUAL: OMS-OMPI-OMC
Chapter II: El contexto normativo para la actuación en materia de innovación y acceso
B. Propiedad intelectual, comercio y otras esferas normativas
Puntos destacados |
|
|
|
|
|
|
|
|
|
|
En la presente sección se ofrece una visión general de los elementos y los instrumentos jurídicos y de actuación relativos a la propiedad intelectual y al sistema de comercio internacional pertinentes para la innovación médica y el acceso a las tecnologías médicas en el ámbito internacional.
1. Sistemas de propiedad intelectual
Se exponen en esta sección las formas de propiedad intelectual más pertinentes para la innovación y el acceso a las tecnologías médicas, así como cuestiones transversales relativas a su aplicación.
(a) Introducción a los sistemas de propiedad intelectual
Estos sistemas funcionan otorgando un derecho limitado a excluir determinados usos, por parte de terceros, de un material protegido. La protección se establece generalmente para reforzar los incentivos comerciales que pueden animar a partes interesadas del sector privado a invertir recursos en la obtención de productos y en la comercialización de nuevas tecnologías. Ese tipo de incentivos cobra especial importancia en el caso del desarrollo de tecnologías médicas, debido a la gran magnitud de los recursos económicos y técnicos requeridos, junto con el elevado riesgo de fracaso, incluso en una fase avanzada del proceso, y con cuestiones derivadas de la responsabilidad por los productos. En muchas tecnologías médicas, el desarrollo es muy costoso, mientras que la reproducción es relativamente económica. En esos casos, no sería viable para las empresas invertir capital en la obtención de un producto y en su autorización reglamentaria, si los competidores pudieran introducir inmediatamente en el mercado una reproducción de ese producto.1
En la medida en que la protección de la propiedad intelectual se basa en el derecho a excluir a terceros, puede llegar a inhibir algunas formas de competencia (tales como la comercialización de medicamentos genéricos) y obstaculizar el avance de la innovación (por ejemplo, cuando no hay una excepción por investigación). La política de propiedad intelectual, las leyes en las que se materializa, y la administración y aplicación de dichas leyes aspiran a equilibrar y reconciliar un abanico de intereses legítimos, buscando una suma positiva que redunde en beneficio del bienestar público.
Los factores de equilibrio son diversos. En el caso de las patentes, consisten en exclusiones de las materias patentables, excepciones y limitaciones a los derechos de patente, limitación del período de protección mediante patente y de los derechos exigibles para mantener en vigor la patente, a fin de facilitar la extinción de patentes infrautilizadas, así como otros instrumentos que superan el ámbito del derecho de patentes, tales como la política de competencia. Si bien los encargados de la formulación de políticas y los legisladores nacionales son quienes determinan en última instancia dónde está el equilibrio, el marco jurídico internacional proporciona el contexto y los principios generales para los sistemas nacionales. El acervo multilateral de normas sobre propiedad intelectual, objeto de esta sección, está determinado por los tratados que administra la OMPI, y por el Acuerdo sobre los ADPIC, que forma parte del sistema jurídico de la OMC e incorpora a su vez las disposiciones sustantivas de varios tratados de la OMPI, entre ellos el Convenio de París (véase el recuadro 2.4).
El Acuerdo sobre los ADPIC tiene importantes repercusiones en la aplicación de la propiedad intelectual a las tecnologías médicas sobre todo gracias a la aplicación de las nuevas normas internacionales que estipulan que las patentes deben poderse obtener para invenciones de cualquier ámbito de la tecnología, en particular los productos farmacéuticos, y que los datos de ensayos clínicos deben protegerse del uso comercial desleal. Las negociaciones del Acuerdo sobre los ADPIC y su ulterior aplicación se han venido centrando en cuestiones de propiedad intelectual y de salud (véase el cuadro 2.3) y, en especial, en la naturaleza y repercusión de las obligaciones establecidas con respecto a las patentes farmacéuticas y la protección de los datos de pruebas.
Recuadro 2.4. El Convenio de París para la Protección de la Propiedad Industrial
|
El Convenio de París se alcanzó en 1883 y se ha revisado en varias ocasiones; la versión más reciente data de 1967. Abierto a todos los Estados, concibe la propiedad industrial en su acepción más amplia, con inclusión de las patentes, las marcas de fábrica o de comercio, los dibujos y modelos industriales, los modelos de utilidad, los nombres comerciales, las indicaciones geográficas y la represión de la competencia desleal. Establece el trato nacional, el derecho de prioridad y las normas comunes. |
El principio de trato nacional en virtud del Convenio de París implica que, en lo que respecta a la protección de la propiedad industrial, cada Estado contratante debe conceder a los nacionales de los demás Estados contratantes las mismas ventajas que concede a sus propios nacionales. Los nacionales de los Estados no contratantes tendrán derecho al trato nacional en determinadas condiciones. |
El derecho de prioridad significa lo siguiente: sobre la base de una solicitud anterior, presentada según el procedimiento normal en uno de los Estados contratantes, el solicitante pide protección para la misma materia objeto de derechos de propiedad industrial por un período de tiempo determinado (plazo de prioridad) en cualquiera de los demás Estados contratantes. Las solicitudes posteriores no se verán afectadas por ningún hecho que pueda haber tenido lugar en el intervalo transcurrido entre la fecha de presentación de la primera solicitud (fecha de prioridad) y la fecha de presentación de la solicitud posterior, como cualquier publicación de la invención reivindicada en una solicitud de patente o venta de artículos que utilicen la marca o en los que esté incorporado el dibujo o modelo industrial. El plazo de prioridad en virtud del Convenio de París es de 12 meses en el caso de las patentes y de los modelos de utilidad, y de seis meses en el caso de los dibujos y modelos industriales y de las marcas. |
Las normas comunes a las que deben atenerse todos los Estados contratantes son las siguientes: |
|
|
|
|
|
|
|
Cuadro 2.3. ADPIC y salud pública: hitos principales |
|
1986 |
En Punta del Este se inauguran las negociaciones de la Ronda Uruguay con un mandato sobre propiedad intelectual. |
1994 |
Concluyen las negociaciones y se adopta el Acuerdo sobre los ADPIC en la Conferencia Ministerial de Marrakech. |
1995 |
Entra en vigor el Acuerdo sobre los ADPIC; se establece la OMC y se le asignan responsabilidades jurídicas y administrativas sobre los ADPIC. |
2000 |
Entran en vigor la mayoría de las obligaciones derivadas de los ADPIC para los Miembros que son países en desarrollo, salvo en el caso de las patentes de productos farmacéuticos. |
2000 |
El grupo especial de la OMC se pronuncia sobre la diferencia en relación con los ADPIC suscitada en torno a las excepciones reglamentarias (excepciones Bolar) para facilitar la introducción de los medicamentos genéricos. |
2001 |
Taller de la OMS y la OMC sobre la fijación diferenciada de precios y financiamiento de medicamentos esenciales (Høsbjør, Noruega). |
2001 |
Declaración de Doha relativa al Acuerdo sobre los ADPIC y la salud pública, con inclusión de la ampliación hasta 2016 del período de transición en la aplicación de la protección de patentes y de datos de pruebas para los países menos adelantados. |
2003 |
Los Miembros de la OMC aprueban el mecanismo del párrafo 6, que permite conceder licencias obligatorias especiales para exportar medicamentos, en forma de flexibilidad adicional prevista en el Acuerdo sobre los ADPIC, inicialmente mediante una exención legal y después mediante el Protocolo de 2005 por el que se enmienda de manera permanente el Acuerdo sobre los ADPIC. |
2005 |
Entra en vigor para los países en desarrollo Miembros de la OMC (pero no para los países menos adelantados) la obligación derivada de los ADPIC de proteger las patentes de productos farmacéuticos. |
2005 |
El Consejo de los ADPIC aprueba la ampliación hasta 2013 del período de transición concedido a los países menos adelantados para aplicar en su totalidad el Acuerdo sobre los ADPIC. |
2010 |
El proceso de examen anual del Consejo de los ADPIC sobre el mecanismo del párrafo 6 aviva el debate de fondo sobre su funcionamiento y aspectos más generales del acceso a los medicamentos. |
El artículo 7 del Acuerdo sobre los ADPIC formula los objetivos de protección y ejercicio de los derechos de propiedad intelectual como un equilibrio entre derechos y obligaciones. Los objetivos hacen referencia a la "promoción de la innovación tecnológica", a la "transferencia y divulgación de tecnología", al beneficio recíproco de los "productores y de los usuarios de conocimientos tecnológicos", así como al "bienestar social y económico". Los principios recogidos en el artículo 8 estipulan explícitamente que los Miembros de la OMC podrán adoptar las medidas necesarias para proteger la salud pública y la nutrición de la población, siempre que esas medidas sean compatibles con lo dispuesto en el Acuerdo sobre los ADPIC. La histórica Declaración de Doha, realizada por la Conferencia Ministerial de la OMC en 2001, ratificó esos objetivos y principios como la orientación adecuada para aplicar lo dispuesto en el Acuerdo en consonancia con las políticas de salud pública. La Declaración hacía referencia a un conjunto de flexibilidades u opciones legales recogidas en el marco de los ADPIC, que se exponen más adelante, después de una presentación general de cuestiones relativas a la propiedad intelectual.
Las normas multilaterales para cada una de las formas de propiedad intelectual son por lo común normas mínimas, que suelen dejar un amplio margen para la aplicación. En el Acuerdo sobre los ADPIC se especifica que los Miembros de la OMC pueden determinar libremente los métodos adecuados para aplicar las normas del Acuerdo en el marco de sus prácticas jurídicas respectivas. Al considerar el abanico de opciones para su aplicación, los encargados de la formulación de políticas tienen en cuenta tanto las normas internacionales como las prácticas de otros países, además de las necesidades y prioridades de su propio país. Los países pueden, si así lo desean, aplicar una protección más amplia, siempre que sea coherente con lo establecido en el Acuerdo. A veces se llama "ADPIC-plus" a esa mayor protección. Un número cada vez mayor de acuerdos bilaterales y regionales establecen normas de ese tipo en las secciones relativas a la propiedad intelectual.2
El principio de no discriminación es una piedra angular del sistema internacional de propiedad intelectual. El "trato nacional" establece que los países no pueden tomar medidas discriminatorias contra nacionales de otros países en el marco de la protección de la propiedad intelectual, salvo lo permitido en excepciones muy concretas. Este principio se formuló ya en 1883, en el texto original del artículo segundo del Convenio de París, y ulteriormente se aplicó en términos generales en el artículo 3 del Acuerdo sobre los ADPIC. El "trato de la nación más favorecida (NMF)" establece que los países no deben adoptar medidas discriminatorias contra nacionales de otros países en relación con la protección de la propiedad intelectual. La aplicación del trato también está sujeta a excepciones. Si bien era una obligación establecida hace mucho tiempo en el derecho mercantil internacional, el trato de la nación más favorecida se aplicó por vez primera a la propiedad intelectual mediante el artículo 4 del Acuerdo sobre los ADPIC. La aplicación de este principio implica que si dos países acuerdan mediante un tratado bilateral otorgar a sus respectivos nacionales un mayor nivel de protección en materia de propiedad intelectual, deben hacer extensivas esas ventajas a los nacionales de los demás Miembros de la OMC.3
Al margen de esos principios generales, cada forma de propiedad intelectual está sujeta a normas concretas, reflejo de los diferentes fines de política que persiguen, de las materias que abarcan y de sus efectos económicos. Esas diferencias se manifiestan en el alcance del objeto de la protección, el alcance de los derechos, la duración de la protección, y la naturaleza de las excepciones y otras salvaguardas por intereses de terceros, así como en el modo en que se hacen cumplir esos derechos.
(b) (b) Legislación y política en materia de patentes
En el último decenio ha habido un incremento considerable del uso de las patentes de tecnologías médicas, que se ha reflejado en el aumento de las solicitudes, en la ampliación del ámbito geográfico de actividad (han aumentado notablemente las patentes de algunas economías emergentes) y en la diversidad de entidades públicas y privadas que solicitan patentes. Ese mismo período ha coincidido con un vivo debate sobre la función del sistema de patentes en relación con la innovación y el acceso a los productos médicos.
En la Declaración de Doha se reconoció el doble efecto de la protección de la propiedad intelectual, a saber, la promoción del desarrollo de nuevos medicamentos y la repercusión en su precio. Desde entonces, el debate se ha centrado en las repercusiones del derecho de patentes sobre el acceso a los medicamentos esenciales. Por otra parte, se ha puesto en tela de juicio si el sistema de patentes ofrece incentivos suficientes y adecuados para facilitar la obtención de nuevos productos en determinadas áreas, por ejemplo, en lo que respecta a las enfermedades desatendidas o a ciertos países. En la práctica, las patentes se utilizan también como medio para concertar diversas asociaciones tecnológicas y colaboraciones en materia de investigación y desarrollo mediante acuerdos de licencias múltiples cuyo objeto es poner las nuevas tecnologías médicas a disposición de la población.
(i) Fundamento del sistema de patentes
La razón de ser de las patentes es hacer que resulte atractivo invertir en innovación y ofrecer un mecanismo para procurar a la sociedad acceso al conocimiento recogido en la solicitud de patente. Entre otras cuestiones, la obligación del titular de la patente de divulgar públicamente su invención permite que la sociedad conozca y, con el tiempo, pueda utilizar, el conocimiento consignado en los documentos de patente. Si una invención pudiera ser libremente utilizada por terceros, sin costo adicional, los "beneficiarios gratuitos" no correrían con ningún gasto derivado del desarrollo. En consecuencia, el inventor original no obtendría los ingresos que había previsto, lo que conduciría, en teoría, a un déficit de invenciones. Según explica la OMPI en un informe reciente, el sistema de patentes tiene como finalidad evitar la deficiencia del mercado derivada del déficit de actividades de innovación, por la razón expuesta. A tal efecto, concede a los innovadores derechos exclusivos limitados que impiden que otros puedan explotar su invención, lo cual facilita que los innovadores puedan obtener un rendimiento adecuado de sus actividades de innovación.4
Sin embargo, el uso del derecho exclusivo puede contribuir a causar una distorsión del mercado y conducir a una situación caracterizada por ineficiencias, precios elevados y déficit de productos. Algunos estudios empíricos realizados muestran que las patentes tienen efectos tanto positivos como negativos sobre la innovación. La falta de datos concluyentes sobre la función del sistema de patentes en el fomento de la investigación y el desarrollo y la transferencia de tecnología impide extraer conclusiones claras acerca de su incidencia en el desarrollo económico.5
Los sistemas de patentes disponen de mecanismos para prevenir y corregir efectos no deseados:
- Los derechos de patente tienen una duración limitada.
- Se permiten exclusiones de la patentabilidad y excepciones y limitaciones a los derechos de patente, a fin de mantener la coherencia con objetivos de política pública más amplios.
- Los procedimientos de solicitud, examen y concesión de patentes, así como la oposición, el recurso y otros procedimientos de revisión dan a los tribunales y a los demás órganos de revisión la posibilidad de corregir decisiones erróneas y proporcionar reparación si es necesario, con el fin de lograr que el sistema de patentes en su conjunto funcione como un instrumento normativo al servicio del interés público.
(ii) El marco internacional
Las normas sustantivas multilaterales para la protección de patentes son principalmente las establecidas en el Convenio de París (Acta de Estocolmo de 1967) y el Acuerdo sobre los ADPIC de 1994. En el primero no se definió qué se considera materia patentable, y hasta la entrada en vigor del segundo, en 1995, hubo una considerable diversidad a ese respecto en la legislación y en la práctica nacionales. En 1988, al principio de las negociaciones sobre los ADPIC, en un informe de la OMPI se citaban 49 países que, o bien no concedían protección mediante patente a los productos farmacéuticos, o bien proporcionaban solamente una protección limitada. Algunos de esos países excluían también los procesos farmacéuticos.6 La duración de las patentes también variaba considerablemente de un país a otro.
El Acuerdo sobre los ADPIC es el primer tratado multilateral que establece los criterios fundamentales7 para la definición de materia patentable. Establece que las patentes deben poder "obtenerse por cualesquiera invenciones, sean de productos o de procedimientos, en todos los campos de la tecnología" (artículo 27 del Acuerdo sobre los ADPIC). La referencia a "todos los campos de la tecnología" significa que pueden obtenerse patentes por productos farmacéuticos (tales como un nuevo compuesto químico con efectos medicinales) y por procesos (tales como un método de fabricación de un medicamento). Establece asimismo que el período de protección conferido no expirará antes de transcurridos 20 años contados desde la fecha de presentación de la solicitud. Esos requisitos entraron en vigor de forma gradual, pero actualmente se aplican a todos los Miembros de la OMC, excepto a los países menos adelantados. La modificación más importante para el área de la salud pública fue la prescripción de que los productos farmacéuticos debían ser patentables en los países en desarrollo a partir de 2005.
A pesar de la existencia de normas internacionales para la protección mediante patentes, no existe lo que se dice una patente de alcance mundial. Las patentes se conceden con arreglo a la legislación nacional, o para el ámbito de una región. El artículo 4bis del Convenio de París establece la independencia de las patentes obtenidas para la misma invención en diferentes países. Es decir, una patente concedida en un país no conlleva derecho alguno en otro país. Una patente concedida para una tecnología farmacéutica en un país concreto no puede utilizarse para evitar la competencia de los medicamentos genéricos en otros países en los que no haya una patente en vigor. Una invención puede patentarse en un país y no en otro.
Recuadro 2.5 El Tratado de Cooperación en materia de Patentes |
|
|
Hay, sin embargo, un sistema mundial para presentar solicitudes de patente: el Tratado de Cooperación en materia de Patentes (PCT), administrado por la OMPI (véase el recuadro 2.5). La decisión final sobre la concesión de la patente carece de alcance internacional; la toman por separado las autoridades nacionales o regionales responsables de las jurisdicciones nacionales en materia de patentes. Por otra parte, varios acuerdos regionales han armonizado y simplificado las leyes en materia de patentes de las distintas regiones.10
A pesar de esta cooperación regional e internacional, la legislación y la práctica en materia de patentes difieren de un país a otro, y pueden dar lugar a resultados divergentes. Cuando las solicitudes de patente para la misma invención se presentan en diferentes oficinas nacionales o regionales de patentes, se procesan por separado de acuerdo con la legislación nacional o regional pertinente, y el resultado final puede no coincidir. Por ejemplo, cuando una solicitud PCT en relación con un compuesto farmacéutico determinado llega a la fase nacional en los Estados contratantes del PCT, puede ocurrir que se apliquen distintos requisitos fundamentales de patentabilidad, en virtud de la ley correspondiente de cada país o región. Sobre la base de la aplicación de esos requisitos en los procesos de examen nacionales, puede suceder que las reivindicaciones hechas en la solicitud de patente11 sean modificadas en un país y se mantengan sin cambios en otro. En consecuencia, la misma solicitud PCT puede dar lugar a una concesión de patente en un país, a una modificación en otro país y al rechazo en un tercer país. Es más, una patente puede ser invalidada por el tribunal de un país, pero confirmada por el tribunal de otro.
La mayoría de las patentes se solicitan y, con el tiempo, se obtienen en un número relativamente pequeño de países, por lo general, aquellos en los que el titular de la patente pretende concentrar los esfuerzos de producción o de comercialización, o en los que hay competidores o capacidad de producción importantes. En los países en los que no se ha presentado una solicitud de patente, o en los que se ha abandonado o denegado una solicitud de patente, la invención reivindicada entra en el dominio público tras la publicación de los documentos de patente, siempre y cuando no haya otra patente u otro derecho sobre la misma tecnología.
(iii) Cuestiones básicas en materia de patentes
Las patentes son derechos territoriales; además, la protección de la patente tiene una duración limitada. Las leyes de patentes proporcionan generalmente un plazo de protección de al menos 20 años. Los titulares de patentes, por otra parte, pueden abandonar una patente antes de agotarse el plazo de protección si, por ejemplo, la comercialización de la invención no genera el rendimiento esperado de la inversión y no llega a cubrir los gastos de mantenimiento de la patente. Las patentes también pueden invalidarse con fundamento en la legislación nacional.
Hay cinco criterios comunes a toda ley de patentes: i) la solicitud debe estar relacionada con la materia patentable; ii) la materia reivindicada debe ser novedosa; iii) debe implicar una actividad inventiva (o no ser evidente); iv) debe ser susceptible de aplicación industrial (o útil) (artículo 27 del Acuerdo sobre los ADPIC); y v) según lo determinado en el artículo 29 del Acuerdo, la invención debe ser debidamente divulgada. Esos requisitos se aplican de forma conjunta; el incumplimiento de cualquiera de ellos conduce al rechazo de la solicitud de patente.
A pesar de que la gran mayoría de los países siguen los mismos criterios fundamentales de patentabilidad, no existe un entendimiento internacional sobre la definición e interpretación de esos criterios. Por ese motivo, existe cierto margen normativo en lo que respecta a su aplicación con arreglo a la legislación nacional pertinente. Las oficinas de patentes y los tribunales interpretan y aplican en cada caso los requisitos nacionales de patentabilidad, dentro del marco jurídico pertinente. Muchas oficinas de patentes proporcionan directrices para el examen de las patentes, con miras a una aplicación uniforme y coherente del derecho de patentes, a menudo basándose en casos que los tribunales competentes han dirimido anteriormente.12
Autoría de la invención, titularidad de la invención y derecho a presentar una solicitud
Toda invención comienza con un inventor o inventores; si bien el derecho internacional en materia de propiedad intelectual no se pronuncia sobre quién debería considerarse el inventor -deja que sean las leyes nacionales las que determinen esa cuestión-, la práctica general es que quienes hayan contribuido a la concepción de al menos una de las reivindicaciones de la patente sean los coinventores, con independencia de la proporción en la que contribuyeron.
La autoría de la invención no entraña necesariamente la titularidad; puede ser que las invenciones realizadas por empleados en el desempeño de su labor, en función de lo que establezca la legislación nacional, pertenezcan a la empresa, haya o no un acuerdo particular al respecto. En los contratos de trabajo o de una empresa de consultoría se podrá establecer que las invenciones realizadas fuera del desempeño laboral de los empleados pertenecen a la empresa o a la parte que contrató al consultor. Frecuentemente, los inventores ceden sus derechos económicos sobre una invención a los organismos que proporcionaron fondos para su investigación.
Las políticas sobre la titularidad de las patentes basadas en investigaciones realizadas en instituciones públicas, tales como las universidades, pueden repercutir de manera significativa en el desarrollo de tecnologías médicas. La falta de directrices claras puede derivar en incertidumbre.
Materia patentable
Las patentes solo se conceden a materias patentables, generalmente definidas como "invenciones" en el derecho de patentes. A falta de una definición de materia patentable consensuada internacionalmente, las leyes nacionales definen los criterios que deben cumplir, ya sea positivamente, ya sea mediante una lista negativa de materias excluidas, o de ambas maneras. Las exclusiones de la materia patentable pueden ser de carácter general; por ejemplo, simples descubrimientos, principios científicos o ideas abstractas. Sin embargo, hay materias patentables que no pertenecen a esas categorías y pueden ser excluidas por otros motivos. Sería el caso, por ejemplo, de las invenciones que se considerarían contrarias a la moral si se explotaran comercialmente (véase el recuadro 2.6), o ciertos métodos para el tratamiento médico de personas o animales (apartado a) del párrafo 3 del artículo 27 del Acuerdo sobre los ADPIC). Algunos países han optado por excluir de la concesión de patentes las invenciones relativas a métodos terapéuticos (o bien, con efecto similar, por limitar el ejercicio de los derechos de esas patentes o por no permitir su ejercicio). Algunas legislaciones nacionales establecen exclusiones muy concretas, como por ejemplo la distinción entre usos médicos primarios y secundarios o, por el contrario, permiten explícitamente la concesión de patentes para tales aplicaciones.19
Recuadro 2.6. Los valores sociales y morales en el sistema de patentes |
|
|
|
|
|
Novedad
El criterio de novedad tiene por objeto que las patentes solo se concedan a tecnologías que aún no están a disposición del público. En muchas jurisdicciones, se entiende por ese criterio que la invención que se reivindica no debe haberse dado a conocer al público, en ningún lugar del mundo, antes de la fecha de presentación o de prioridad de la solicitud de patente; por ejemplo, mediante una publicación o por haberse realizado, presentado oralmente o usado ante el público, antes de presentar la solicitud. Las leyes nacionales definen qué tipo y forma de documentación constituye, si procede, una divulgación previa al público, pertinente para la evaluación de la novedad.
Por ejemplo, imaginemos un caso en el que una solicitud de patente reivindica un nuevo tipo de férula para inmovilizar el brazo de un paciente. En el momento en que se presentó la solicitud solo los empleados de la empresa conocían la invención. Los empleados no podían, en virtud de la obligación contraída en su contrato de trabajo, divulgar su conocimiento al público. No obstante, si antes de presentar la solicitud se probó la férula en pacientes, sin que se hubieran convenido y aplicado los acuerdos de confidencialidad pertinentes, es posible que la invención que se reivindica no pueda considerarse novedosa puesto que el acceso al conocimiento correspondiente no se restringió lo suficiente y, en consecuencia, podría considerarse que ya se ha dado a conocer al público.
Actividad inventiva o carácter no evidente
El derecho de patentes, en general, solo define el concepto básico de lo que constituye actividad inventiva y deja la interpretación a las oficinas de patentes y a los tribunales de supervisión. En la práctica, se han ideado distintos métodos para determinar la existencia de una actividad inventiva sobre la base de determinados indicadores, comprobados por un examinador de patentes. En muchas jurisdicciones, ese criterio se entiende en el sentido de que la invención debe aportar un avance técnico suficiente con respecto a la situación hasta ese momento -es decir, un avance técnico sobre lo que se ha utilizado o se ha descrito hasta ese momento en la esfera en cuestión- que no resultaría evidente una persona con una experiencia o un nivel de conocimientos medio que trabajara en el área técnica a la que pertenece la invención (una "persona del oficio"). Por ejemplo, la actividad inventiva (o el carácter no evidente) se pueden basar en un efecto "inesperado" o "sorprendente" que no resultaría evidente, en el momento de la invención, para una persona familiarizada con esa área de la tecnología. La consideración de evidente o no evidente puede variar con el tiempo. A guisa de ejemplo: a fines del siglo XX, aislar un gen suponía un arduo trabajo; hoy en día, sin embargo, es mucho más corriente.20
Aplicación industrial o utilidad
La aplicación industrial o utilidad significa que la invención puede ser producida o utilizada en cualquier sector industrial, incluido el agropecuario, o que tiene una utilidad concreta, creíble y considerable. En general, la aplicación de ese requisito no plantea problemas prácticos. En el área de la biotecnología, sin embargo, el asunto no está tan claro; las solicitudes de patente que reivindican invenciones relacionadas con genes podrían bloquear el uso de la secuencia génica reivindicada para usos que el solicitante desconocía en el momento de la solicitud. No estaría justificado, por lo tanto, conceder una patente en relación con una función de la que el solicitante ni siquiera era consciente.21
Divulgación
Para obtener la concesión de una patente es necesario divulgar suficientemente la invención. El artículo 29 del Acuerdo sobre los ADPIC dispone que el solicitante de una patente debe divulgar la invención de manera suficientemente clara y completa para que las personas capacitadas en la técnica de que se trate puedan llevar a efecto la invención. En algunos países, se exige al solicitante que indique también la mejor manera de llevar a efecto la invención que conozca el inventor en la fecha de presentación de la solicitud. Asimismo, es posible que el solicitante deba revelar detalles sobre las patentes solicitadas o concedidas en otras jurisdicciones.
Algunos críticos del sistema de patentes sostienen que, a menudo, la divulgación de una invención no es suficiente para "fabricar" la materia patentada. Una de las cuestiones fundamentales que se plantean en relación con el requisito de divulgación es en qué medida debe el titular de una patente revelar su invención, en el marco del sistema de patentes, a fin de contribuir a la promoción de la innovación tecnológica y a la transferencia y difusión de la tecnología, en beneficio recíproco de los productores y de los usuarios de conocimientos tecnológicos. En una patente, la invención debe describirse de manera que una persona capacitada en la técnica de que se trate pueda realizar la invención sin demasiada experimentación o excesivos intentos. Sin embargo, para producir la invención de manera rentable, la información técnica contenida en la patente a menudo debe complementarse con información que se considera al alcance del lector especializado de la patente. El requisito de divulgación fue concebido para satisfacer fines jurídicos y técnicos específicos del sistema de patentes. La información técnica difundida mediante este sistema no puede sustituir a otras fuentes de información, por ejemplo, los libros de texto y las publicaciones científicas.22
En algunos casos, es posible que por inadvertencia se conceda una patente que no ha cumplido con el requisito de una divulgación suficiente según la ley regional o nacional pertinente. En ese caso, la patente podría ser defectuosa. La mayoría de las leyes sobre patentes establecen procedimientos para la revocación o invalidación de patentes que no cumplan con los requisitos reglamentarios de patentabilidad. Así pues, sería una estrategia arriesgada para el titular de una patente optar intencionadamente por divulgar una invención de manera insuficiente, que no satisfaga el requisito de divulgación según la legislación nacional o regional pertinente.23
(iv) Procedimiento en materia de patentes
Por lo general, compete a la oficina de patentes receptora de la solicitud establecer si la invención reivindicada cumple con todos los criterios de patentabilidad. En algunos países, la oficina nacional o regional de patentes realiza una búsqueda relativa al estado de la técnica24 y un examen de fondo. Si la oficina establece que se han cumplido todos los requisitos del caso, se concede la patente. El examen de fondo permite obtener un mayor grado de seguridad jurídica sobre la validez de las patentes concedidas, superior a la seguridad que proporcionaría un sistema que se limitara a registrar las solicitudes sin llevar a cabo tal examen.
Sin embargo, si la calidad del trabajo de búsqueda y del examen es deficiente, ello puede resultar perjudicial porque se crean falsas expectativas sobre la validez de la patente. Por otra parte, si las oficinas de patentes carecen de los recursos necesarios para mantener al día la documentación relativa al estado de la técnica, y para emplear a examinadores con la experiencia necesaria -o si no reciben un número suficiente de solicitudes que justifique contar con examinadores calificados en todas las áreas técnicas-, puede ser que un sistema basado en exámenes de fondo no sea el método más apropiado. Hay opciones, tales como la concesión de patentes sin realizarse un examen de fondo; el registro de patentes concedidas tras haberse realizado un examen de fondo en algún otro lugar; el uso de búsquedas y resultados de exámenes realizados por otras oficinas de patentes; y la cooperación entre oficinas de patentes.25 Por ejemplo, el Tratado de Cooperación en materia de Patentes proporciona búsquedas internacionales y exámenes internacionales preliminares no vinculantes, realizados por una serie de oficinas de patentes designadas a tal efecto por la Asamblea de la Unión Internacional de Cooperación en materia de Patentes. Las oficinas nacionales de patentes pueden utilizar esos informes de búsqueda y examen para decidir si conceden o no una patente.
Actualmente, algunos países desarrollados y en desarrollo emplean "sistemas de registro" (en contraposición a los "sistemas de examen"), que no contemplan realizar un examen de fondo y, por lo tanto, no juzgan si la invención reivindicada satisface o no los criterios de patentabilidad. Se ha argumentado que es sensato aplazar el examen de fondo hasta que la patente en cuestión sea realmente objeto de litigio. La validez de ese argumento puede depender del costo, la duración y la cantidad de litigios sobre patentes, por una parte, y del costo que suponen la creación y el mantenimiento de un sistema de examen, por otra parte. En países donde el sistema judicial no funciona del todo bien puede ser difícil rectificar las patentes concedidas erróneamente.
Cuando el derecho de patentes estipula un examen completo de las solicitudes, las oficinas de patentes las analizan con arreglo a los criterios de patentabilidad oficiales y sustantivos. Como resultado, a menudo los solicitantes se ven en la necesidad de reducir el alcance de sus reivindicaciones para evitar que sus solicitudes sean rechazadas. Asimismo, pueden tener que retirar las reivindicaciones que el examinador de patentes considere que no cumplen con los criterios de patentabilidad, bien sea porque ya se conocen y no resultan novedosas o porque son obvias y no implican una actividad inventiva. El alcance de los derechos recogidos finalmente en una patente es a menudo muy inferior a lo que se reivindicaba inicialmente en la solicitud.26
(v) Procedimientos de examen
En la práctica, puede suceder que una patente se conceda erróneamente; para hacer frente a esta situación, los sistemas de patente incluyen procedimientos de examen (ante un órgano administrativo, como por ejemplo una junta de apelaciones, o ante un tribunal). En algunos países, terceras partes pueden oponerse a la concesión de una patente ante un órgano administrativo, en un plazo limitado. Lo anterior complementa los procedimientos que sigue la oficina para conceder patentes y permite que el público contribuya a velar por su calidad. Algunos países establecen procedimientos de oposición previa a la concesión; otros, procedimientos de oposición posteriores a la concesión; incluso los hay que establecen ambos.27
(vi) Derechos que confiere una patente
Una vez concedidas, las patentes confieren a sus titulares el derecho de impedir que cualquier tercero fabrique, use, ofrezca para la venta, venda o importe la invención patentada en el país en el que se ha concedido el derecho de patente (artículo 28 del Acuerdo sobre los ADPIC). El alcance de la protección conferida por la patente se determina sobre la base de las reivindicaciones de esta. Las reivindicaciones deben redactarse de manera clara y concisa, justificarse plenamente mediante la información aportada sobre la invención.
En la práctica, las patentes se utilizan no solo para excluir a los competidores, sino también para permitir que otros fabriquen, usen, ofrezcan para la venta, vendan o importen la invención patentada, por medio de licencias.
Los titulares pueden otorgar licencias sobre sus patentes, o vender o transferir su titularidad. Una licencia es un contrato en que el titular autoriza a otra persona a utilizar su propiedad intelectual, ya sea a cambio de un pago de regalías (o alguna otra consideración) o de forma gratuita, en un ámbito y un territorio determinados (por un período que puede abarcar toda la vida de la patente). Las licencias se utilizan con frecuencia para permitir que otras empresas con conocimientos técnicos especializados en investigación o desarrollo tengan acceso al conjunto diverso de tecnologías patentadas necesarias para fabricar un producto farmacéutico complejo, en condiciones convenidas de mutuo acuerdo.28
Las patentes y las autorizaciones para la venta son cuestiones independientes. La concesión de una patente sobre un medicamento nuevo en un país no confiere a su titular el derecho a vender el medicamento en ese país sin la aprobación de la autoridad de reglamentación competente. Por otra parte, que una patente haya sido concedida o no tiene nada que ver con su aprobación reglamentaria. Algunos países, sin embargo, estipulan que quien solicite una aprobación reglamentaria debe presentar información sobre la concesión o no de las patentes, y no permiten que las autoridades de reglamentación autoricen la venta mientras siga vigente una patente pertinente ("autorización de comercialización o vinculación de patentes").29
((vii) Excepciones y limitaciones
Las excepciones y limitaciones a los derechos de patente son herramientas, comunes a todos los sistemas de propiedad intelectual, que se utilizan para abordar intereses divergentes. Permiten, por ejemplo, restringir ciertos usos de la invención patentada en el ejercicio de los derechos de patente. En los artículos 5 y 5ter del Convenio de París se establecen normas acerca de las licencias obligatorias y ciertas limitaciones de los derechos exclusivos, con miras a salvaguardar el interés público. En los artículos 30 y 31 del Acuerdo sobre los ADPIC se prevén excepciones y limitaciones a los derechos, así como las condiciones en que se podrán aplicar.30
Una excepción muy común es la excepción por investigación, que permite a terceros utilizar la invención patentada con fines de investigación, durante la vida de la patente.31 Igualmente frecuente es la excepción basada en el examen reglamentario, que permite a los fabricantes de productos genéricos competidores hacer un uso limitado de una invención patentada antes de que venza la protección de la patente, para obtener la autorización de comercialización de un producto competidor. Se conoce también como la excepción Bolar, y se aborda en el subapartado i) del apartado a) de la sección C.3 del capítulo IV.
Las leyes nacionales también podrán, en determinadas condiciones, conceder "licencias obligatorias" a terceros, para su propio uso o para uso de los gobiernos o en nombre de estos, sin la autorización del titular de la patente. En virtud de una licencia obligatoria y la autorización de su utilización por los gobiernos, un tribunal o autoridad competente otorga permiso expreso a una persona distinta del titular de la patente para producir, importar, vender o usar el producto protegido mediante patente, o para utilizar el proceso protegido mediante patente, para hacer frente a necesidades concretas. Los titulares de la patente tienen derecho a percibir una remuneración. El Acuerdo sobre los ADPIC establece una serie de requisitos relativos a la forma en que deben emitirse las licencias obligatorias y la autorización de su utilización por los gobiernos, con el fin de definir algunos límites prácticos y así salvaguardar algunos intereses del titular. En particular, cada caso será considerado en función de sus circunstancias propias (apartado a) del artículo 31); será necesario haber intentado previamente negociar una licencia voluntaria, excepto en circunstancias de extrema urgencia o de uso público no comercial (apartado b) del artículo 31); y la licencia se limitará principalmente al abastecimiento del mercado interno (apartado f) del artículo 31). Hay limitaciones sobre el alcance y la duración de esa autorización (apartado c) del artículo 31), así como sobre su retirada (apartado g) del artículo 31). El derecho de uso de la patente será no exclusivo (apartado d) del artículo 31); y no podrá cederse a terceros (apartado e) del artículo 31). El titular tiene derecho a solicitar un examen judicial o administrativo que podría conducir a la retirada de la autorización de uso o licencia (apartado g) del artículo 31) y dar derecho a recibir una remuneración adecuada (apartado h) del artículo 31).
Se podrá eximir de la obligación de negociar una licencia voluntaria en un plazo prudencial cuando concurran situaciones de emergencia nacional, otras circunstancias de extrema urgencia o en casos de uso público no comercial (apartado b) del artículo 31). Cuando se haya autorizado el uso de una patente sin consentimiento de su titular, para corregir prácticas anticompetitivas y tras un proceso judicial o administrativo, los Miembros de la OMC no están obligados a aplicar las condiciones expuestas. En esos casos, la licencia no tiene por qué ser principalmente para el abastecimiento del mercado interno (se permite por tanto la exportación en cantidades no limitadas) y la cuantía de la remuneración puede ser diferente (es decir, puede tratarse de una cuantía inferior o incluso no remunerarse en absoluto). Algunos países han recurrido a las licencias obligatorias y a la utilización por los gobiernos a fin de producir o importar productos farmacéuticos de fabricantes genéricos a menor precio, con el fin de aumentar el acceso a los medicamentos, antes de que expiren las patentes correspondientes.31
La limitación de destinar las licencias obligatorias y la utilización por el gobierno principalmente al abastecimiento del mercado interno, establecida en el apartado f) del artículo 31 del Acuerdo sobre los ADPIC, fue revisada tras la Declaración de Doha con el objeto de permitir, en circunstancias concretas, la producción exclusivamente con fines de exportación en virtud de una licencia obligatoria (véase el subapartado ii) del apartado a) de la sección C.3 del capítulo IV).
(viii) Información sobre patentes
El sistema de patentes exige la divulgación pública de las invenciones y convierte las patentes publicadas (y las solicitudes de patente, en muchos países) en una importante fuente de información técnica y jurídica. La información sobre patentes ofrece una base para las estrategias y las decisiones comerciales y relativas a la propiedad intelectual, así como un punto de partida para los procesos de investigación y desarrollo. El sistema de patentes constituye en sí mismo un registro, exhaustivo y sistemático, de conocimientos técnicos (Bregonje, 2005).33
Las normas, recomendaciones y directrices de la OMPI ayudan a las oficinas de propiedad industrial a crear y administrar sus sistemas de información y publicación de patentes.34 Gracias a ello, la estructura de los documentos sobre patentes es bastante uniforme en todo el mundo. Las normas abordan la transmisión, el intercambio, la comunicación y la difusión de información sobre patentes entre las oficinas de propiedad industrial, y facilitan el acceso a información técnica y su recuperación.35 La búsqueda de información sobre patentes resulta así más fácil y accesible.
No obstante, la forma en que se publican las patentes varía considerablemente de un país a otro. Según el artículo 12 del Convenio de París, las oficinas de patentes deben publicar regularmente los nombres de los titulares de las patentes concedidas, con una breve designación de las invenciones patentadas, en una hoja oficial periódica. En la práctica, las solicitudes de patente se publican de forma accesible al público generalmente 18 meses después de la fecha de presentación (fecha de prioridad). De manera similar, el artículo 21 del Tratado de cooperación en materia de patentes (PCT) exige de forma general que las solicitudes internacionales PCT se publiquen transcurridos 18 meses a partir de la fecha de prioridad. Algunos países publican solamente las patentes concedidas, y no las solicitudes de patente. La publicación puede limitarse a una breve nota sobre la concesión de la patente, en cuyo caso el acceso a la información técnica y a la evaluación del alcance y la situación jurídica de una patente es mucho más difícil, y solo una inspección de archivos en la oficina de patentes permitirá obtener información detallada sobre la invención reivindicada. Los países también pueden optar por publicar información adicional de utilidad, como por ejemplo informes de búsqueda y examen, correcciones, modificaciones, traducciones e información sobre la situación jurídica.
La familia de patentes hace referencia a varios documentos de patente relacionados entre sí mediante uno o varios documentos de prioridad comunes o técnicamente equivalentes. Las solicitudes realizadas posteriormente en otros países suelen reivindicar la prioridad de la primera solicitud. Así pues, los integrantes de una familia de patentes pueden estar relacionados entre sí mediante una reivindicación de prioridad. Dado que en las presentaciones ulteriores se pueden reivindicar varias prioridades, correspondientes a solicitudes anteriores, hay distintos conceptos de familia.36 En las bases de datos no siempre se utiliza la misma definición de familia de patentes y por tal motivo es posible que las búsquedas por familias arrojen resultados diferentes según la base que se consulte.
Gracias a la publicación y la digitalización de la información sobre patentes, el conocimiento es ahora más accesible y fácil de consultar. Sin embargo, la recuperación, el análisis y la explotación de la información sobre patentes son cuestiones muy complejas y requieren aptitudes especializadas. Asimismo, realizar búsquedas eficaces puede plantear problemas en relación con la disponibilidad de la información en las bases de datos (OMPI, 2010).
(ix) Situación de la patente e información sobre su situación jurídica
La situación de la patente y la información sobre su situación jurídica ayudan a determinar la libertad para operar con respecto a un proyecto y en qué medida y con quién hay que negociar las licencias. El término "situación de la patente" se utiliza en el presente estudio para referirse a todas las patentes relacionadas con un producto determinado, mientras que el término "situación jurídica" hace referencia a los actos jurídicos y administrativos que se producen durante el ciclo de vida de una única patente.37
Todos los registros de patentes recogen los actos jurídicos más importantes, tales como la concesión de la patente y su titularidad. Solo de esas fuentes primarias puede obtenerse información fiable y autorizada sobre la situación jurídica de una patente. Las fuentes secundarias también pueden proporcionar información, a menudo con un cierto retraso, pero es posible que carezcan de algunos datos que sí contienen las fuentes primarias.38
Evaluar la situación de las patentes de productos médicos suele requerir conocimientos específicos. Un producto (incluidos los medicamentos fabricados mediante asociación de componentes, como por ejemplo las asociaciones de dosis fijas), su proceso de fabricación y su uso pueden estar cubiertos por varias patentes, que protegen diversos aspectos tecnológicos. Los fabricantes y vendedores de un producto no están obligados a revelar todas las patentes pertinentes. Además, es complicado verificar la situación jurídica de todos los integrantes de una familia de patentes.
En el caso de los medicamentos comercializados en los Estados Unidos, se puede obtener información en el Orange Book39 de la Administración de Alimentos y Medicamentos (FDA), donde se enumeran los medicamentos aprobados por dicho organismo y se ofrece información sobre las patentes para productos conexos y modos de utilización. No se incluyen las patentes de procedimiento ni las que reivindican embalajes, metabolitos y productos intermedios. La información sobre esas patentes no se presenta a la FDA.40 El Ministerio de Salud del Canadá tiene un registro de patentes similar, donde se recoge una lista por orden alfabética de componentes de medicamentos, las patentes conexas, las fechas de caducidad y otra información afin.41 Medicines Patent Pool ha puesto a disposición del público en una base de datos la información relativa a la situación jurídica de las patentes sobre los medicamentos antirretrovíricos (véase el recuadro 2.7).
(x) Tendencias de presentación de solicitudes en virtud del sistema del Tratado de Cooperación en materia de Patentes (PCT)
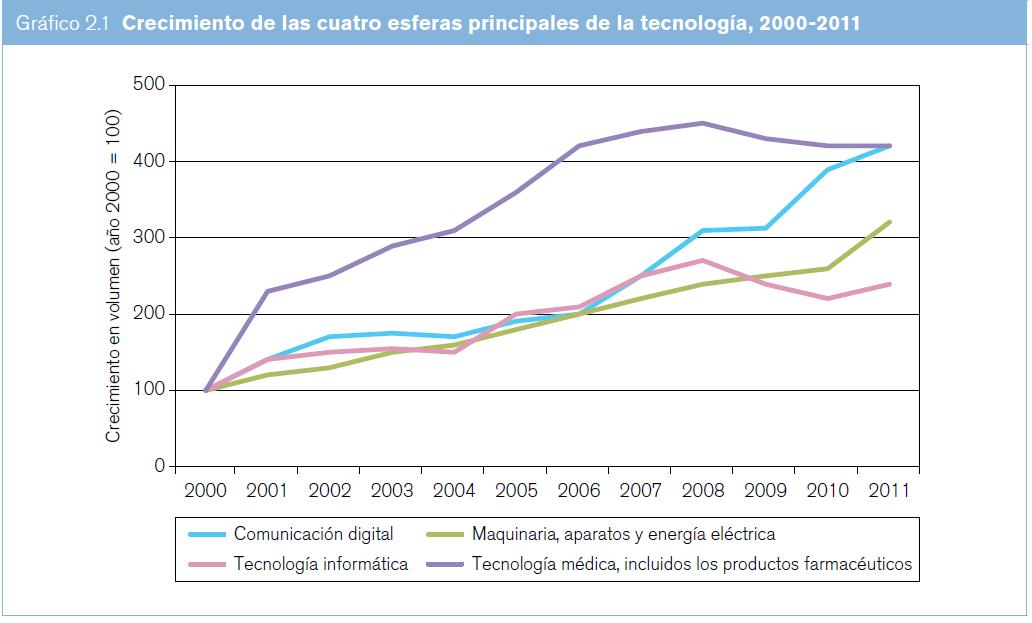
Según la OMPI (2012), la mayoría de las solicitudes PCT presentadas entre 1978 y 2011 pertenecían al ámbito de la tecnología médica. Sin embargo, representan una proporción relativamente pequeña del total de solicitudes (6,6% en 2011). Cabe señalar que el término "tecnologías médicas", tal como se utiliza por la OMPI (2012), no se corresponde con el sentido que se le da en el presente estudio, donde se incluyen bajo esa denominación los datos relativos a los productos farmacéuticos (4,7% de las solicitudes PCT presentadas en 2011). Así, las solicitudes PCT relativas tanto a tecnologías médicas como a productos farmacéuticos representaron el 11,3% de las solicitudes presentadas en 2011 y, en combinación, las tecnologías médicas y los productos farmacéuticos representan el campo de la tecnología con mayor número de solicitudes PCT presentadas entre 1978 y 2011 (véase el gráfico 2.1).
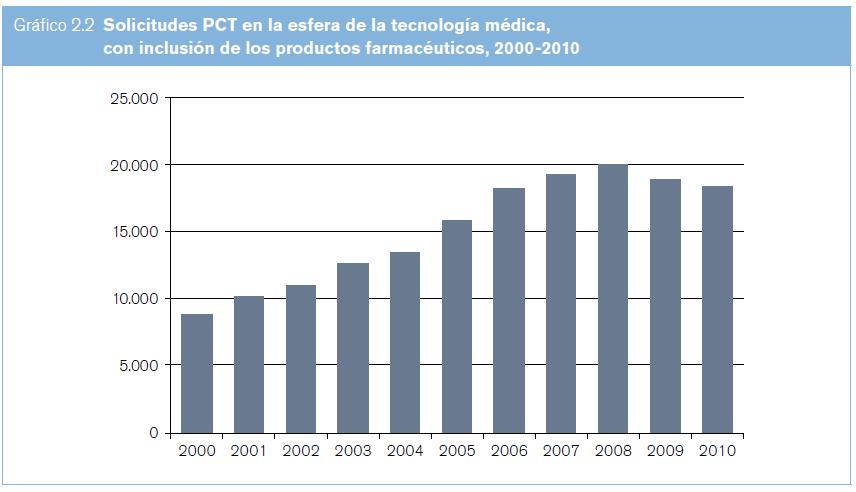
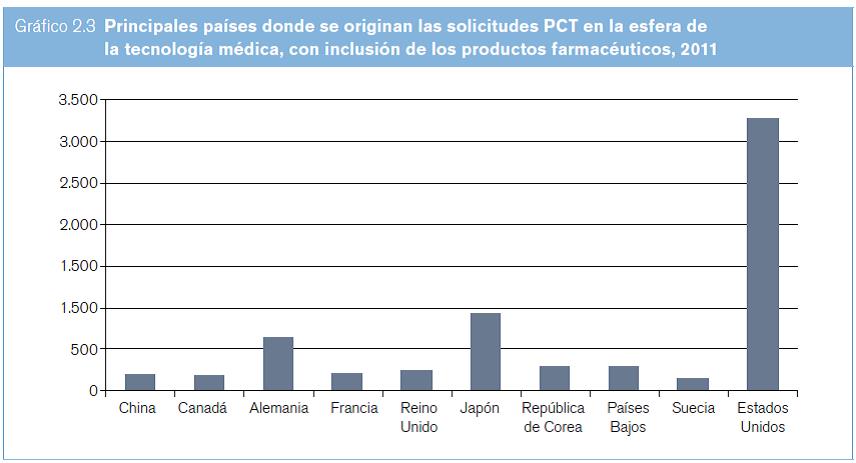
Entre los años 2000 y 2010, la cantidad anual de solicitudes PCT presentadas en el área de las tecnologías médicas se mantuvo entre 4.496 y 10.481; en la de los productos farmacéuticos, entre 3.789 y 7.863. En lo que respecta a las tecnologías médicas (según se entiende en el presente estudio, es decir, con inclusión de los productos farmacéuticos), la cantidad anual de solicitudes PCT presentadas entre los años 2000 y 2010 se mantuvo entre 8.785 y 18.344 (véase el gráfico 2.2). Las cantidades totales aumentaron cada año hasta 2008, y en los dos años siguientes disminuyeron. Entre los 10 países de origen principales figuran los Estados Unidos, el Japón, la República de Corea y varios países de Europa occidental (véase el gráfico 2.3).
Recuadro 2.7. Base de datos de Medicines Patent Pool sobre la situación de las patentes de determinados medicamentos contra la infección por el VIH |
Medicines Patent Pool ha creado una base de datos sobre la situación de las patentes de determinados antirretrovíricos en algunos PBI y PIM. Los datos sobre la situación jurídica se han obtenido y contrastado utilizando diversas fuentes, en particular las oficinas nacionales y regionales de patentes, que facilitaron la información por conducto de la OMPI. Si bien la información procede de fuentes primarias, la base de datos proporciona únicamente una instantánea correspondiente a un momento determinado, e incluye solo algunas de las patentes relativas a cada antirretrovírico. Se muestra la fecha de caducidad prevista, sobre la base de un plazo de 20 años a partir de la fecha de presentación de la solicitud. Sin embargo, es posible que algunas patentes hayan expirado o vencido, o que hayan sido retiradas, rechazadas, modificadas, revocadas o impugnadas, después de la inclusión de la información correspondiente en la base de datos. Así pues, si más adelante se necesita información precisa, es importante verificar la vigencia de la información ante la autoridad competente.42 |
Fuente: Base de datos estadísticos de la OMPI.
Fuente: Base de datos estadísticos de la OMPI.
(c) Ensayos clínicos y protección de los datos de pruebas
Tal como se ha expuesto en la sección A.6 del capítulo II, en los países donde se realiza una evaluación independiente de la calidad, inocuidad y eficacia de los medicamentos, los organismos de reglamentación exigen generalmente la presentación de datos de pruebas para obtener la autorización de comercialización de un producto farmacéutico nuevo. Compete a la empresa solicitante (no a las autoridades públicas) generar los datos de pruebas farmacológicas, toxicológicas y clínicas. La protección de los datos de pruebas afecta al uso que los organismos de reglamentación pueden hacer de los datos confidenciales recogidos en el expediente de solicitud del fabricante del producto originario; lo que guarda una estrecha relación con la reglamentación farmacéutica. Al mismo tiempo, es parte del sistema de propiedad intelectual, ya que representa una forma de protección contra la competencia desleal. La razón fundamental de proteger los datos es que, con frecuencia, se necesita realizar una inversión considerable, tanto en tiempo como en dinero, para obtener los datos, especialmente ante unos requisitos normativos cada vez más estrictos. Al generar los datos, por lo tanto, las empresas fabricantes de los productos originarios tienen gran interés en proteger su inversión. Por el contrario, los intereses públicos en pugna pueden estar tratando de facilitar el acceso a los productos genéricos. Por esa razón, la protección de los datos de pruebas es uno de los temas más polémicos en el debate sobre salud pública y propiedad intelectual.
Fuente: Base de datos estadísticos de la OMPI.
(i) Normas jurídicas internacionales
El artículo 10bis del Convenio de París (que exige una protección eficaz contra la competencia desleal en general) y, en particular, el Acuerdo de la OMC sobre los ADPIC, contienen normas multilaterales sobre esta cuestión.
El Acuerdo establece que los Miembros de la OMC deberán impedir la divulgación no autorizada y el uso comercial desleal de la información confidencial presentada a un organismo de reglamentación, con arreglo a ciertas condiciones. Se protegerán los datos de prueba contra:
- La divulgación: se trata de la obligación directa de no divulgar los datos presentados a efectos de obtener la autorización reglamentaria. Los organismos de reglamentación pueden, sin embargo, divulgar los datos cuando sea necesario para proteger al público, o cuando se adopten medidas para garantizar la protección de los datos contra todo uso comercial desleal (véase el recuadro 3.6 del capítulo III).
- El uso comercial desleal: en el Acuerdo sobre los ADPIC no se define el término "uso comercial desleal" ni se aborda el modo de lograr dicha protección. Por lo tanto, difieren las opiniones y las prácticas de los países sobre lo que se exige exactamente. Algunos sostienen que la manera más eficaz de garantizar esa protección es conceder a las empresas fabricantes de productos originarios un plazo razonable de exclusividad de los datos. En el marco de tal régimen, durante un número de años determinado los organismos de reglamentación pertinentes no podrían basarse en los datos presentados en la solicitud de autorización del producto originario para aprobar ulteriores versiones genéricas del producto, posiblemente apoyadas en datos de bioequivalencia indicativos de que dicho producto genérico es similar o fundamentalmente similar al originario. Otros no están de acuerdo con la opinión de que el Acuerdo sobre los ADPIC exija exclusividad, y sostienen que hay otras formas lícitas de protección contra el uso comercial desleal. En las negociaciones de la Ronda Uruguay se debatió la posibilidad de hacer de la exclusividad de los datos una obligación explícita en el marco del Acuerdo; los negociadores, sin embargo, aprobaron el texto general del párrafo 3 del artículo 39 en su forma actual.
No existe jurisprudencia ni orientación autorizada de la OMC sobre ninguna de esas cuestiones (el tema se planteó, aunque no se resolvió, en las consultas celebradas entre los Estados Unidos y la Argentina en el marco del mecanismo de solución de diferencias de la OMC; la solución que alcanzaron de mutuo acuerdo se limitó a señalar que las partes habían expresado sus puntos de vista y habían acordado que las diferencias en las interpretaciones serían resueltas sobre la base de las reglas del Entendimiento sobre Solución de Diferencias (véanse los documentos de la OMC WT/DS171/3 y WT/DS196/4)). Tampoco se resolvieron en el Consejo de los ADPIC celebrado como preparación de la Conferencia Ministerial de Doha en 2001, aunque los Miembros presentaron algunos puntos de vista sobre la interpretación del párrafo 3 del artículo 39 del Acuerdo sobre los ADPIC. Lo que sí puede afirmarse, sin embargo, es que: i) las flexibilidades y la interpretación favorable a los intereses de la salud pública que figura en la Declaración abarca la totalidad del Acuerdo y, por tanto, se aplican a la protección de datos de pruebas en virtud del párrafo 3 del artículo 39; ii) no hay ninguna prescripción ADPIC que establezca explícitamente la obligación de proporcionar la exclusividad de los datos, pero sí se exige algún tipo de protección contra el uso comercial desleal; y iii) el hecho de que en virtud del párrafo 3 del artículo 39 del Acuerdo sobre los ADPIC se deban proporcionar dos formas de protección pone de relieve que la protección contra el uso comercial desleal no debe limitarse simplemente a no divulgar los datos.
Con todo, deben cumplirse ciertas condiciones para que se pueda aplicar la protección de los datos de pruebas, a saber:
- Los datos no deben haber sido divulgados: el párrafo 3 del artículo 39 solo exige la protección de los datos no divulgados, es decir, de la información que no se ha publicado anteriormente. Si los datos ya han sido divulgados, por ejemplo, en una revista científica, un documento de patente o algún otro medio, no es necesario velar por su protección.
- La presentación de datos de pruebas debe ser una exigencia de los países: los países que no exijan la presentación de datos de pruebas u otros datos para llevar a cabo su propio examen reglamentario sobre un producto farmacéutico no están obligados en virtud del Acuerdo a proteger los datos de pruebas con respecto a ese producto. La obligación de proteger los datos se deriva solamente de la existencia de un requisito reglamentario de presentar los datos como condición para obtener la autorización de comercialización.
- Los productos para los que se solicita la autorización de comercialización han de contener entidades químicas novedosas: el Acuerdo sobre los ADPIC solo hace referencia a los datos de pruebas incluidos en las solicitudes de autorización de comercialización de productos que utilizan "entidades químicas novedosas". No incluye una definición de este término, y la OMC no ha determinado su ámbito de aplicación.
- La obtención de los datos debe exigir un esfuerzo considerable: el Acuerdo no especifica la naturaleza de ese esfuerzo, es decir, si debe ser de carácter técnico o económico. Tampoco establece que el solicitante esté obligado a demostrar que se ha realizado tal esfuerzo.
En cualquier caso, los países menos adelantados Miembros de la OMC no están obligados a proteger los datos de pruebas relativos a productos farmacéuticos, debido a la ampliación del período de transición, establecido actualmente hasta el 1º de enero de 2016.
(ii) La distinción entre protección mediante patentes y protección de datos de pruebas
Las patentes y los datos de pruebas son dos categorías distintas de la esfera de la propiedad intelectual. El Acuerdo sobre los ADPIC prevé la protección de los datos de pruebas como una forma de protección contra la competencia desleal en la sección correspondiente a la protección de la información no divulgada, y no en la sección correspondiente a las patentes. Una patente protege una invención -por ejemplo, una nueva molécula- con independencia del esfuerzo o de la inversión que conlleva, mientras que la protección de los datos de pruebas abarca una materia diferente: concretamente, la información presentada para solicitar la autorización reglamentaria (en ocasiones llamada "expediente de registro"). Así pues, puede suceder que una patente sea titularidad de una parte, y el expediente reglamentario, de otra (por ejemplo, del licenciatario local de una patente). Ambas formas de protección pueden ir paralelas en el caso de los medicamentos patentados que consiguen llegar al mercado. Sin embargo, lo normal es que la protección de la patente comience unos años antes porque la solicitud correspondiente suele presentarse inmediatamente después de obtenerse la invención, mientras que los ensayos clínicos no se realizan hasta una etapa posterior del ciclo de obtención del producto. En el momento en que comienzan dichos ensayos, la patente puede estar aún pendiente o haberse concedido ya. Dado que la protección de los datos de pruebas y la protección de las patentes son dos cuestiones distintas, es posible que proteger los datos de pruebas conlleve ciertas ventajas para la empresa que los ha generado. Sería así, por ejemplo, cuando el producto en cuestión no está protegido mediante patente, cuando no le queda más que un corto período de protección o cuando se ha impugnado la validez de una patente en un procedimiento de oposición. En esas situaciones, la existencia de un período de exclusividad puede retrasar la introducción de los genéricos en el mercado, ya que los fabricantes de genéricos están obligados a esperar a que expire dicho período.
(iii) Aplicación en los países
La discrepancia antes mencionada en cuanto a la manera de proteger los datos de pruebas en el marco del Acuerdo sobre los ADPIC se refleja también en la forma en que se incorpora esta obligación a las legislaciones nacionales. En consonancia con sus prioridades políticas, los países han adoptado diferentes enfoques para lograr la protección contra el uso comercial desleal. En muchos casos, el modo elegido se ha guiado también por determinadas disposiciones suscritas en el marco de un acuerdo de libre comercio (ALC)43 o, a veces, mediante el compromiso jurídicamente vinculante de prever expresamente la exclusividad de los datos de pruebas en los protocolos de adhesión a la OMC (es el caso de China y Ucrania, por ejemplo). Esos países han acordado así asumir obligaciones más concretas que las que se exigen en el Acuerdo sobre los ADPIC.
La mayoría de los países desarrollados, y algunos países en desarrollo, prevén un régimen de exclusividad de los datos. Otros, como la India y muchos otros países en desarrollo, prohíben a sus autoridades de reglamentación permitir a terceros el acceso y el uso de la información que reciban, con arreglo a la legislación sobre confidencialidad y competencia desleal. Sin embargo, no prohíben a las autoridades de reglamentación basarse en los datos de pruebas presentados en la solicitud de registro de un producto originario ya aprobado para examinar y aprobar las solicitudes de ulteriores aspirantes a entrar en el mercado. Además, no conceden un período de exclusividad.
Hay otras opciones que se proponen para la protección de datos de pruebas, como los modelos de compensación o de participación en los gastos, en virtud de los cuales se permitiría utilizar los datos del producto originario, a condición de que el proveedor del producto genérico participe en los costos de obtención de los datos. Los Estados Unidos, por ejemplo, prevén tanto la exclusividad de los datos como un sistema obligatorio de retribución por información en relación con los datos presentados en las solicitudes de autorización reglamentaria de plaguicidas (pero no para los productos farmacéuticos). El acuerdo de libre comercio entre la Asociación Europea de Libre Comercio y Corea (artículo 3, anexo XIII) también admite un sistema de compensación como alternativa a la exclusividad de los datos.
Los países que conceden derechos de exclusividad suelen establecer un período fijo de entre 5 y 10 años, con posibilidad de ampliación en algunos casos. Por lo general, el período comienza a partir de la fecha de autorización de venta del producto originario en el mismo país en que se solicita la protección de los datos de pruebas. Algunos Miembros de la OMC, como la Unión Europea y los Estados Unidos, conceden un período adicional de exclusividad en el caso de nuevas indicaciones y formulaciones.
En algunos países se establecen ciertas excepciones y limitaciones a la exclusividad de los datos. La ley de los Estados Unidos reduce el período de exclusividad a cuatro años cuando el solicitante de un segundo producto certifica que la patente es nula o que el segundo producto no infringe la patente (condicionado a una posible suspensión en el marco de una acción por la infracción de una patente). El Canadá no prevé la exclusividad de los datos si el producto originario no se comercializa en su territorio. Tampoco lo hacen Chile ni Colombia, si el producto originario no se vende en sus respectivos territorios dentro de los 12 meses siguientes a la concesión de la autorización de venta local. Chile no prevé exclusividad de los datos si la solicitud de autorización de venta local se presenta más de 12 meses después de la concesión inicial del registro o la autorización de venta en otro país.
Hay otras excepciones basadas en la protección del interés público, como por ejemplo en situaciones de emergencia sanitaria o en el marco de las exportaciones bajo licencia obligatoria en virtud del sistema previsto en el párrafo 6.44 Si adopta la forma de exclusividad de los datos, la protección de los datos de pruebas podría obstaculizar la aplicación de las licencias obligatorias de patentes, incluso cuando un país exija el examen reglamentario de los productos destinados a la exportación en virtud del sistema previsto en el párrafo 6.45 El Canadá y la Unión Europea decidieron no aplicar la protección de los datos en el caso de los productos fabricados bajo licencia obligatoria con fines exclusivamente de exportación en virtud del sistema mencionado. Chile no prevé la exclusividad de los datos si el producto está sujeto a una licencia obligatoria, del tipo que sea.
(iv) Las dimensiones de innovación y acceso de la protección de los datos de pruebas
El modo en que se protegen esos datos resulta muy pertinente para el fomento de la innovación de productos y de la mejora del acceso a las tecnologías médicas existentes. La forma que adopte la protección en el ámbito nacional influirá en la obtención o introducción de nuevos productos, y determinará la prontitud con la que empiece la competencia entre un producto originario y otro genérico.
A efectos de su aprobación reglamentaria, los medicamentos nuevos deben someterse a varias fases de ensayos clínicos a fin de comprobar su inocuidad y eficacia. Esos requisitos reglamentarios forman parte integral del proceso de obtención de cualquier producto médico nuevo, lo que distingue la innovación en medicina de la de otras áreas tecnológicas. En la actualidad, la obtención de datos sobre calidad, inocuidad y eficacia mediante ensayos clínicos sigue estando financiada -a pesar de las diversas propuestas y los debates en torno a esta cuestión- principalmente por empresas que aspiran a introducir en el mercado una nueva tecnología médica.
A pesar de que los ensayos clínicos responden a objetivos sanitarios legítimos, los costos que acarrean suponen un importante obstáculo para la introducción en el mercado de nuevos productos farmacéuticos. Dado que las solicitudes de patente sobre los compuestos químicos se presentan por lo general en una etapa relativamente temprana del proceso de investigación y desarrollo de un producto, la larga duración de los ensayos clínicos, junto con el pertinente proceso de aprobación reglamentaria, reducen de hecho el período de exclusividad comercial del producto patentado, lo que reduce el margen para recuperar los costos de investigación y desarrollo del producto en cuestión, y de otros productos fallidos.
Por esa razón, la industria farmacéutica de investigación sostiene que la protección de los datos de pruebas -y, en especial, la exclusividad de los datos-, proporciona un importante incentivo para que la industria invierta en la obtención de nuevos productos y en los ensayos clínicos conexos. Además, las empresas innovadoras, obviamente, aprecian la relativa certeza de la exclusividad de los datos, sobre todo en comparación con la incertidumbre asociada a la validez o el alcance de una posible patente, lo que, a su vez, provoca incertidumbre sobre la posibilidad de dejar temporalmente al margen a los competidores. Ejemplo de ello sería la obtención de la versión pediátrica de un medicamento ya existente, que en algunas jurisdicciones no da lugar a patente por no considerarse novedosa. En una situación así, la protección de los datos de pruebas clínicas sería el único incentivo para invertir en la obtención de ese producto. Algo similar podría suceder en relación con los ensayos clínicos para someter a prueba la inocuidad y la eficacia de medicamentos tradicionales conocidos que no son patentables por no ser novedosos.
Por otra parte, los defensores de la sanidad pública subrayan que, en lo que respecta a los países en desarrollo, el incentivo adicional para realizar investigación y ensayos clínicos es mínimo, mientras que el efecto negativo sobre los precios, y, por lo tanto, sobre el acceso a las tecnologías médicas, es considerable. De manera similar, el Grupo Consultivo de Expertos en Investigación y Desarrollo: Financiación y Coordinación (GCEID), en un informe publicado en abril de 2012, consideró que "no había pruebas de que la exclusividad de los datos contribuyese sustancialmente a la innovación con respecto a las enfermedades de los tipos II y III y a las necesidades específicas de I+D de los países en desarrollo en relación con las enfermedades de tipo I; por consiguiente, concluimos que su eliminación, en caso de producirse, no afectaría negativamente a los incentivos a la innovación con respecto a estas enfermedades y además contribuiría a reducir los precios de los medicamentos conexos" (OMS, 2012a).
Uno de los aspectos principales con respecto al acceso a los medicamentos es la forma de abordar la cuestión de las solicitudes de autorización de comercialización de productos genéricos idénticos. En un régimen de exclusividad de los datos, la introducción en el mercado de los medicamentos genéricos puede verse retrasada, ya que los ulteriores solicitantes tienen que esperar a que venza el período de exclusividad. El fabricante del producto genérico podría, en principio, hacer los ensayos clínicos de nuevo o acordar el uso de los datos originales con el fabricante del producto originario, pero en la práctica no sucede así. Ello es debido, entre otras razones, al costo y el tiempo que conlleva la obligación de obtener los datos de pruebas. Por el contrario, los ulteriores aspirantes al mercado del mismo medicamento pueden evitar tener que presentar datos originales si se les permite utilizar los datos que el fabricante originario presentó en su solicitud para demostrar que sus productos tienen un efecto equivalente (bioequivalencia). De esa manera, los productos genéricos competidores pueden acceder antes al mercado, ya sea cuando no hay protección mediante patente o bien cuando esta ha vencido. Así, al permitirse la comercialización de un medicamento competidor, los consumidores disponen de una alternativa y, por lo general, se reducen los precios. Desde la perspectiva de la salud pública, esto se considera positivo porque evita la duplicación de los ensayos clínicos, contraria a la ética, y acelera la introducción de los medicamentos genéricos en el mercado. Sin embargo, desde el punto de vista del primer solicitante puede considerarse injusto, porque los ulteriores aspirantes al mercado no se ven obligados a invertir en costosos ensayos clínicos (algunos de los cuales pueden resultar fallidos) y, por tanto, podrían competir directamente a un costo considerablemente menor.
La cuestión de la protección de los datos de pruebas es una buena muestra del dilema fundamental al que se enfrenta la protección de la propiedad intelectual. Con el objeto de incentivar la obtención de nuevos productos, en algunos países se establece explícitamente un período de exclusividad comercial para facilitar la obtención del rendimiento de la inversión, a pesar de que con ello se puede retrasar la entrada de los genéricos en el mercado.
(v) Productos biosimilares: protección de los datos de pruebas farmacológicas, toxicológicas y clínicas
Una cuestión que cobra una importancia cada vez mayor, y que repercute tanto en los sistemas de innovación como en el acceso a la nueva generación de medicamentos "biológicos", es la relativa a la protección de los datos de pruebas farmacológicas, toxicológicas y clínicas presentados a un organismo de reglamentación para respaldar la autorización de productos originarios de referencia. Los modelos establecidos para la protección de los productos farmacéuticos clásicos de moléculas pequeñas no son necesariamente apropiados para los medicamentos biológicos, más complejos y difíciles de reproducir (véase el recuadro 2.3 sobre los productos biosimilares). En Suiza y en la Unión Europea, entre otros, la exclusividad de los datos asociada a la protección de datos de pruebas farmacológicas, toxicológicas y clínicas se aplica tanto a los medicamentos de moléculas pequeñas como a los productos bioterapéuticos. Aunque la directiva 2004/27/CE,46 prevé la presentación de datos suplementarios en el caso de los medicamentos biológicos, que son distintos de los medicamentos genéricos, no establece normas específicas para la exclusividad de los datos de ese tipo de productos. Por lo tanto, se aplican las normas para la autorización de medicamentos genéricos.
Por el contrario, el Congreso de los Estados Unidos aprobó una legislación específica mediante la Ley de competencia de precios e innovación en los productos biológicos, de 2009. La FDA puede no aprobar la solicitud de un producto biosimilar hasta transcurridos 12 años desde la fecha en que se autorizó por primera vez el producto de referencia. La duración de la exclusividad para los productos biológicos es distinta de la concedida a los medicamentos de moléculas pequeñas o a los medicamentos "huérfanos" (poco rentables) que, según la ley de los Estados Unidos, es de solo cinco y siete años, respectivamente.
(d) Marcas de fábrica o de comercio
(i) El sistema de marcas de fábrica o de comercio
Las marcas de fábrica o de comercio permiten a las empresas fabricantes y comercializadoras diferenciar sus productos de los de sus competidores. Ayudan a los consumidores a elegir con conocimiento de causa, y su objetivo es evitar que estos sean engañados. El registro de marcas de fábrica o de comercio está sujeto a ciertos requisitos, relativamente estandarizados en todo el mundo y recogidos en prácticamente todos los derechos de marcas. Esas marcas tienen que ser distintivas, o al menos tener potencial para llegar a serlo, de los bienes o servicios de su titular y no deben inducir a error. No deben infringir derechos adquiridos por terceros ni estarán formadas exclusivamente por signos o indicaciones que puedan servir, en el comercio, para designar la especie, la calidad, la cantidad, el destino, el valor, el lugar de origen de los productos o la época de producción, o que hayan llegado a ser usuales en el lenguaje corriente o en las costumbres constantes. Los términos genéricos que utilizan palabras comunes para definir la categoría o el tipo de producto no son distintivos, y todos los competidores deben poder utilizarlos sin tener que observar derechos de marca de fábrica o de comercio.
Hay una diferencia crucial entre la denominación genérica de un producto -por ejemplo, ampicilina-, que debe poder usarse para identificar cualquier producto pertinente, y la marca de fábrica o de comercio registrada que utiliza una empresa para diferenciar el producto de cuya fabricación y distribución es responsable. A estas últimas a veces se las llama "nombres comerciales". La OMS mantiene un registro sistemático de los nombres genéricos, las llamadas denominaciones comunes internacionales (DCI), universalmente reconocidas como denominaciones únicas que identifican determinadas sustancias farmacéuticas o principios activos farmacéuticos. Las marcas de fábrica o de comercio están vinculadas a un producto y tanto la industria farmacéutica de investigación como la de productos genéricos las utilizan para crear confianza y establecer una relación entre la empresa, el médico que prescribe y el paciente, lo que puede permitir a los propietarios de marcas de fábrica o de comercio cobrar precios más altos. Con frecuencia se distingue entre los fabricantes farmacéuticos "de marca" y los fabricantes farmacéuticos genéricos, pero esta distinción puede inducir a error, ya que unos y otros utilizan marcas de fábrica o de comercio para comercializar y diferenciar sus productos.
Las marcas de fábrica o de comercio se protegen con arreglo a las leyes de cada país o región, no a escala mundial. Todos los países signatarios del Convenio de París tienen un registro de esas marcas. Las solicitudes de registro de marcas deben presentarse por separado en cada país o región donde se desee utilizarlas, o ante la OMPI, utilizando el Sistema de Madrid para el Registro Internacional de Marcas (véase el recuadro 2.8).47 No es extraño que una marca esté protegida en algunos países y no en otros.
Recuadro 2.8. El Sistema de Madrid para el Registro Internacional de Marcas |
El Sistema de Madrid se rige por el Arreglo de Madrid relativo al Registro Internacional de Marcas (pactado en 1891) y por el Protocolo sobre el Arreglo de Madrid relativo al Registro Internacional de Marcas (pactado en 1989). El Sistema de Madrid ofrece a los titulares de las marcas un modo sencillo, flexible y accesible de obtener protección para su marca en mercados exteriores, y de mantener esa protección. Mediante la presentación de una solicitud internacional en un idioma (inglés, francés o español), y el pago de las tasas en una única moneda (francos suizos), el titular de una marca puede obtener protección en más de 80 países, incluida la Unión Europea, siempre y cuando cuente con un "registro de base", es decir, una solicitud o registro de marca de fábrica o de comercio presentada en una "oficina de origen". La Oficina Internacional de la OMPI realiza un examen de forma; pero cualquier cuestión de fondo, como por ejemplo si la marca reúne las condiciones para la protección o si está en conflicto con una marca anterior, se deja a criterio de cada Parte Contratante designada, con arreglo a su legislación nacional sobre el particular. Si la oficina de marcas de una Parte Contratante designada no deniega la protección en un plazo determinado, la protección de la marca de fábrica o de comercio es la misma que si se hubiera registrado en la oficina correspondiente. |
Por otra parte, el Sistema de Madrid simplifica en gran medida la gestión de la marca, ya que implica un único registro internacional, con una única fecha de renovación a tener en cuenta, y permite conseguir protección en muchas Partes Contratantes designadas. Posteriormente, se puede ampliar la protección de la marca a otras Partes Contratantes. El registro internacional también se puede renovar y modificar. Las modificaciones podrían consistir por ejemplo en cambios en el nombre o en la dirección del titular, o en un cambio de titularidad, y pueden realizarse siguiendo un procedimiento centralizado. |
La Oficina Internacional inscribe la marca en el Registro Internacional, publica el registro internacional en la Gaceta de la OMPI de Marcas Internacionales, y envía la notificación correspondiente a cada Parte Contratante designada. |
El titular de una marca de fábrica o de comercio tiene el derecho exclusivo de impedir que otros usen signos que sean idénticos o similares a su marca en determinados tipos de bienes o servicios cuando dicha utilización pueda dar lugar a confusión. El titular de una marca y, por lo general, cualquier licenciatario, pueden hacer valer sus derechos contra posibles infracciones. Estas marcas no tienen un plazo máximo de protección; pueden renovarse indefinidamente, siempre y cuando permanezcan en uso y mantengan su carácter distintivo. El derecho a una marca se puede perder por anulación o eliminación del registro, si la marca no se renueva o no se abonan las tasas de renovación. Asimismo, puede perder su carácter distintivo y convertirse en un término genérico, lo cual puede suceder si el titular o el público, con el consentimiento de este, la utiliza como denominación genérica de un producto o como un término de uso común. Las normas mínimas internacionales para la protección de las marcas de fábrica o de comercio se recogen en el Convenio de París y en el Acuerdo sobre los ADPIC.
(ii) Las marcas de fábrica y de comercio y las denominaciones comunes internacionales (DCI)
A diferencia de las marcas de fábrica o de comercio, que constituyen derechos exclusivos, las DCI son nombres genéricos de los principios activos farmacéuticos. Cada DCI es un nombre único, reconocido a escala mundial en casi todos los Estados miembros de la OMS y no está sujeto a derechos exclusivos. La OMS tiene el mandato constitucional de desarrollar, establecer y promover normas internacionales con respecto a productos biológicos, farmacéuticos y similares. La secretaría de la OMS y el Grupo de Expertos en DCI de la OMS colaboran estrechamente con los comités nacionales de nomenclatura, los organismos de reglamentación farmacéutica, las farmacopeas y la industria farmacéutica para seleccionar un nombre único aceptable en todo el mundo para cada principio activo que se vaya a comercializar como producto farmacéutico.
Es importante que haya una nomenclatura internacional, en este caso las DCI, para los principios farmacéuticos porque facilita que los medicamentos se puedan identificar inequívocamente y recetar y dispensar de forma segura, además de facilitar la comunicación y el intercambio de información entre profesionales de la salud y científicos de todo el mundo. Como nombres únicos, las DCI deben ser distintivas tanto fonética como ortográficamente, y no deben ser susceptibles de confundirse con otros nombres de uso común. A fin de que las DCI se conozcan, la OMS las pone oficialmente en el dominio público, de ahí la designación de "comunes". Cualquier productor o distribuidor puede utilizar una DCI en su producto, siempre y cuando la utilice con precisión. "Ibuprofeno", por ejemplo, es una DCI, y cualquier productor o distribuidor puede utilizarla para designar su producto.
Otra característica importante del sistema DCI es que las relaciones químicas y farmacológicas de los principios activos relacionados entre sí se hacen patentes mediante un "formante" común que forma parte del nombre. El empleo de formantes comunes permite que los médicos, los farmacéuticos o cualquier persona que maneje productos farmacéuticos sepan que un principio activo determinado pertenece a un grupo de sustancias con actividad farmacológica similar. Por ejemplo, todos los anticuerpos monoclonales reciben el sufijo o formante "-mab", mientras que todos los antagonistas adrenérgicos tienen como sufijo o formante "-olol".
Es importante que las marcas de fábrica o de comercio se distingan claramente de las DCI, con vistas a la identificación inequívoca de los productos y, por ende, a la seguridad de los pacientes. Es asimismo imprescindible mantener las DCI en el dominio público y no conceder derechos de propiedad privada para su empleo. Las marcas de fábrica o de comercio no deben derivarse de las DCI y, en especial, no deben incluir los formantes comunes. Usar un formante común en una marca registrada dificultaría seriamente la selección ulterior de nombres dentro de esa serie. Por esa misma razón, las DCI no deben construirse sobre la base de marcas de fábrica o de comercio ya existentes. En ese sentido, el Grupo de Expertos en DCI convocado por la OMS suele rechazar las propuestas de DCI que contengan una marca conocida, y hay un procedimiento para abordar las objeciones de las partes interesadas, que pueden estar basadas en la similitud entre la propuesta de DCI y una marca. Por otra parte, las marcas que incluyen un formante oficial infringen el sistema DCI. La OMS ha pedido a los Estados miembros que no concedan marcas de fábrica o de comercio, ni ningún otro derecho de propiedad exclusivo, a denominaciones comunes internacionales o sus formantes. Cuando se publica una lista de DCI propuestas o recomendadas, la OMS la distribuye a todos sus Estados miembros. Las listas se pueden consultar en el sitio Web de la OMS.48
A raíz de una decisión del Comité Permanente sobre el Derecho de Marcas, Dibujos y Modelos Industriales e Indicaciones Geográficas (SCT), y en colaboración con el Programa de DCI, la OMPI notifica oficialmente a las oficinas nacionales y regionales de marcas de sus Estados miembros la publicación de cada nueva lista de DCI propuestas y recomendadas. Una encuesta realizada por la OMPI en las oficinas de marcas mostró que el 72% de las oficinas encuestadas examinaban las solicitudes de marca de fábrica o de comercio para detectar posibles conflictos con las DCI.49
Distinguir claramente la DCI de la marca de fábrica o de comercio registrada facilita la selección de medicamentos en los procesos de adquisición pública. La razón es que abordar la compra de un producto bajo su denominación común internacional abre el proceso a todos los fabricantes del producto designado de esa manera. Muchos países exigen un etiquetado inequívoco que muestre la DCI en una posición separada tanto del nombre de la empresa fabricante, ya sea de productos genéricos u originarios, como del nombre comercial o de la marca de fábrica o de comercio. Si bien el Acuerdo sobre los ADPIC establece que no se complicará el uso de una marca de fábrica o de comercio en el curso de operaciones comerciales con exigencias especiales, permite algunas limitaciones justificables en el artículo 20. El etiquetado erróneo o engañoso también puede considerarse competencia desleal; está regulado por el artículo 10bis del Convenio de París, así como por las leyes para la protección del consumidor y otras disposiciones similares de diversos países que pretenden prevenir esa práctica.
(iii) Autorización reglamentaria de los nombres comerciales
El nombre con que se va a comercializar un nuevo medicamento (es decir, la marca de fábrica o de comercio o el nombre comercial) también está sujeto al examen de las autoridades de reglamentación; su aprobación forma parte de la autorización de venta. La similitud entre los nombres de algunos medicamentos y los errores de medicación que se produjeron en los años noventa llevaron a la Administración de Alimentos y Medicamentos (FDA) de los Estados Unidos y a la Agencia Europea de Medicamentos (EMA) a establecer el examen de las denominaciones registradas, a fin de proteger la salud y la seguridad de las personas.50 En el último decenio, el examen de esos nombres como parte de la autorización reglamentaria ha cobrado un carácter más oficial al crearse órganos especializados en la FDA y en la EMA,51 que rechazan del 30% al 40% de los nombres comerciales presentados para su registro.52
Los criterios que aplican los organismos de reglamentación farmacéuticas para evaluar los nombres comerciales pretenden hacer frente a confusiones y posibles errores de medicación en el contexto específico de las prácticas de distribución y prescripción farmacéutica. Así pues, coinciden en parte con los criterios que se examinan en el contexto de las solicitudes de marca de fábrica o de comercio. Su objetivo es excluir los nombres que contengan o impliquen alusiones a la eficacia y la inocuidad del medicamento que sean falsas, engañosas o no avaladas mediante datos. Además, teniendo en cuenta los riesgos relativos a la prescripción farmacéutica, el examen reglamentario descarta los nombres que en su forma verbal o escrita sean similares a otros nombres de medicamentos o a las abreviaturas que se suelen utilizar en las recetas escritas a mano, tales como pautas de dosificación, formas farmacéuticas o vías de administración.
El requisito de autorización del nombre comercial de los nuevos medicamentos, dentro de la autorización reglamentaria general de los productos farmacéuticos, es importante para velar por la inocuidad en el ámbito de la distribución y la prescripción farmacéuticas. Dado que los organismos pertinentes autorizan la comercialización de los medicamentos bajo un nombre determinado (es decir, no pueden venderse bajo otro nombre), las compañías farmacéuticas tienen que dar con un nombre que, además de merecer la aprobación oficial, pueda ser marca de fábrica o de comercio protegida en los principales mercados donde se vaya a vender el producto. A fin de alcanzar ese doble objetivo, y para asegurarse el éxito, normalmente las empresas se inventan varios nombres posibles para cada medicamento nuevo, y los registran como marcas de fábrica o de comercio en sus mercados principales antes de presentarlos como distintas opciones a los organismos de reglamentación. Esa práctica explica en parte la proliferación de solicitudes de marca de fábrica o de comercio en la esfera de los productos farmacéuticos, que en 2010 representaron el 4,7% de las solicitudes de marca (OMPI, 2011a). Esa profusión de solicitudes puede conducir a una abundancia de marcas registradas pero en desuso.
(e) Derechos de autor y productos farmacéuticos
Los derechos de autor atañen a toda creación original del ámbito artístico o literario, con independencia del tipo de trabajo (tal y como se establece en la Convención de Berna para la protección de las obras literarias y artísticas y se recoge en el Acuerdo sobre los ADPIC), pero no abarca ideas, procedimientos, métodos de operación ni conceptos matemáticos en sí.
En el caso de los productos farmacéuticos, una cuestión determinante en relación con los derechos de autor es si la protección abarca o no los prospectos o folletos informativos que acompañan a dichos productos. Los fabricantes de productos genéricos pueden utilizar la información que figura en un prospecto, ya que los derechos de autor no abarcan esa información como tal, solo la forma en que se ha expresado. Sin embargo, dado que la protección de esos derechos abarca por lo general la realización de copias de obras originales a escala comercial, en ocasiones los tribunales han determinado que los productores de medicamentos genéricos no pueden utilizar en sus productos copias directas de las expresiones originales que figuran en los prospectos del producto originario. Así se determinó en 2002 en Sudáfrica, en relación con el prospecto del antibiótico amoxicilina/clavulanato de potasio.53En 2011 se tomó en un principio una decisión similar en Australia, en relación con la leflunomida, un medicamento para la artritis reumatoide. El Tribunal Federal dictaminó que los derechos de autor sobre los documentos de información del producto seguían vigentes. Sin embargo, posteriormente, el Parlamento de Australia aprobó en 2011 una enmienda a la Ley de Derechos de Autor por la cual se establecía que el uso -en cualquier forma, incluso mediante reproducción directa- de la información de producto ya autorizada para otros productos farmacéuticos no constituye una infracción de los derechos de autor. Una resolución judicial ulterior confirmó que las empresas fabricantes de productos genéricos pueden reproducir la información aprobada por la Administración de Productos Terapéuticos, sin infringir por ello los derechos de autor en una variedad de circunstancias determinadas.54
(f) Observancia
El valor de las normas de propiedad intelectual que se han detallado más arriba depende de la existencia de un sistema de ejecución eficaz. Dado que los derechos de propiedad intelectual son derechos privados, por lo general la responsabilidad de su aplicación recae sobre los propios titulares. Por lo tanto, cuando se produce una infracción son normalmente los titulares quienes entablan una acción civil. Sin embargo, es el interés público lo que está en juego cuando la infracción de la propiedad intelectual se produce en el ámbito penal; por ejemplo, cuando un comerciante, sin permiso y a sabiendas, fabrica, distribuye o vende, a escala comercial, productos con la marca de fábrica o de comercio de otra empresa. Dicho esto, la aplicación de los derechos de propiedad intelectual es una cuestión claramente separada de la reglamentación de medicamentos con vistas a su seguridad, calidad y eficacia, incluida toda medida contra los productos médicos de calidad subestándar, espurios, de etiquetado engañoso, falsificados o de imitación (CSEEEFI).
(i) Los vínculos entre la aplicación de los derechos de propiedad intelectual y la salud pública
El significado de "falsificación" es diferente en el contexto de la salud pública y en el de la propiedad intelectual, y también lo es la motivación para luchar contra los productos médicos CSEEEFI, en el terreno de la salud pública y en el de la propiedad intelectual.55 Desde la perspectiva de la salud pública, motivada por aspectos reglamentarios, el término "falsificación" se utiliza en su sentido más amplio, y no debe confundirse con la infracción de la marca. Por esa razón, en los debates sobre políticas de salud ha sido sustituido por el término recién mencionado.56 La lucha contra los productos médicos CSEEEFI está motivada exclusivamente por la amenaza a la salud pública y por otras preocupaciones conexas relativas a la protección del consumidor. Desde la perspectiva de la propiedad intelectual, la condición determinante para considerar que un producto es falsificado es la utilización comercial de una marca de fábrica o de comercio sin la autorización de su titular. Por lo tanto, el objetivo es preservar los intereses del titular de la marca en el ejercicio de sus derechos y proteger el interés público luchando contra las infracciones cuando se producen en el ámbito penal.
Si bien la motivación puede ser diferente, los métodos utilizados para prohibir la producción, el comercio y la distribución de todo tipo de productos que infringen la marca y de los productos médicos CSEEEFI tienen algunas semejanzas; los más comunes son los controles aduaneros y el derecho penal. Por ejemplo, los productos farmacéuticos figuran con frecuencia entre los principales productos suspendidos por las autoridades aduaneras por infringir los derechos de propiedad intelectual.57 La razón es que, en el comercio internacional, las marcas de fábrica o de comercio juegan un papel importante como identificadores comerciales e indicadores del origen comercial y pueden ayudar a detectar productos falsificados. Los falsificadores utilizan sin autorización las marcas para hacer creer, con frecuencia aunque no siempre, que el producto es auténtico, manifestando en falso su identidad y origen. En ese sentido, algunos sostienen que las medidas de observancia de la propiedad intelectual para luchar contra la falsificación de marcas pueden tener efectos secundarios positivos consistentes en mantener productos peligrosos fuera del mercado. Otros argumentan que las consideraciones de salud pública deben mantenerse claramente separadas de las cuestiones de observancia de la propiedad intelectual, con vistas a mantener netamente delimitados los objetivos de fomentar la observancia de los derechos privados y de trabajar en beneficio de la salud pública, sobre todo si se tiene en cuenta la retención de productos genéricos en tránsito por Europa.58
(ii) Observancia en el marco del Acuerdo sobre los ADPIC
El Acuerdo sobre los ADPIC establece el único marco multilateral completo para hacer valer los derechos de propiedad intelectual. Contiene un conjunto de normas mínimas que protegen los derechos de los titulares de propiedad intelectual sin obstaculizar el comercio legítimo. Esas normas incluyen procedimientos judiciales y vías de recurso que deben ponerse a disposición de los interesados, como los mandamientos judiciales, las demandas por perjuicios y las órdenes de destrucción de los bienes que infringen una marca. Esas vías de recurso deben ser aplicables a todos los derechos que abarca el Acuerdo, como son las patentes, la protección de los datos de pruebas, las marcas de fábrica o de comercio y los derechos de autor. Los procedimientos administrativos, tales como las actuaciones ante las autoridades administrativas, son facultativos y tienen que ajustarse a los principios por los que se rigen los procedimientos civiles. En el caso de las mercancías de marca de fábrica o de comercio falsificadas, tal y como se definen en el Acuerdo, incluidos los productos médicos, y en el de las mercancías pirata que lesionan el derecho de autor, se debe ofrecer una gama más amplia de procedimientos, tales como medidas aduaneras y procedimientos penales. El Acuerdo incluye asimismo ciertas obligaciones generales o normas de actuación que estipulan que los Miembros de la OMC deben velar por que esos procedimientos concretos de ejecución permitan adoptar medidas eficaces, con inclusión de recursos ágiles para prevenir e impedir las infracciones. Esos procedimientos se aplicarán de forma que se evite la creación de obstáculos al comercio legítimo, y deberán prever salvaguardias contra su abuso. En el Acuerdo se aclara que los Miembros de la OMC no están sujetos a obligación alguna con respecto a la distribución de los recursos entre los medios destinados a lograr la observancia de los derechos de propiedad intelectual y los destinados a la observancia de la legislación en general.59
(g) Los elementos de flexibilidad previstos en el Acuerdo sobre los ADPIC y la Declaración de Doha
Determinar cuál es la mejor opción para un país, dentro de la gama de posibilidades de que dispone, es una cuestión fundamental en la concepción del régimen nacional de propiedad intelectual. Sin embargo, muchas de esas posibilidades normativas, a las que se suele llamar "flexibilidades previstas en el Acuerdo sobre los ADPIC", forman parte de los mecanismos utilizados en los sistemas de patentes para mantener un equilibrio entre los intereses públicos y los privados, desde mucho antes de que se negociara el Acuerdo y antes de que se formulara la Declaración.
(i) La flexibilidad en el sistema de propiedad intelectual
La adopción de las normas del Acuerdo sobre los ADPIC se tradujo en una gama de opciones que los Miembros de la OMC podían adoptar para cumplir con las obligaciones contraídas, teniendo en cuenta distintas consideraciones como el grado de desarrollo del país y sus intereses nacionales particulares (por ejemplo, en materia de salud pública). Sin embargo, a pesar de las frecuentes referencias a la "flexibilidades" en los debates sobre políticas, ni el Acuerdo ni ninguno de los instrumentos ulteriores han definido oficialmente el significado exacto del término, que en el propio Acuerdo se utiliza muy poco. De hecho, a pesar de que el uso de la flexibilidad puede ser mucho más amplio, tanto para los países en desarrollo como para los países desarrollados, se hace referencia explícita a la "flexibilidad" exclusivamente en relación con las necesidades especiales de los Miembros que son países menos adelantados para establecer una base tecnológica sólida y viable, con lo que se explica la razón del período de transición adicional concedido a esos países (véanse el Preámbulo y el párrafo 1 del artículo 66 del Acuerdo sobre los ADPIC). La expresión "flexibilidades" solo se introdujo en el vocabulario de los círculos interesados en la propiedad intelectual en la preparación de la Declaración de Doha, y, sobre todo, tras la conclusión de las negociaciones correspondientes.60
Al integrar la función de las "flexibilidades", la Declaración aclaró la importancia de las decisiones concretas adoptadas por cada país en la aplicación del Acuerdo y dio un mayor relieve a aquellas. Ello obedece a la importancia fundamental que adquirió el debate sobre las distintas medidas políticas para promover la salud pública desde que se inició la labor preparatoria de las negociaciones de Doha, que culminarían en 2001 con la adopción de la Declaración. El Acuerdo pone de relieve la existencia de elementos de flexibilidad o "flexibilidades" y su importancia para el sector farmacéutico, y la Declaración confirma "el derecho de los Miembros de la OMC de utilizar, al máximo, las disposiciones del Acuerdo sobre los ADPIC, que prevén flexibilidad" para proteger la salud pública. En la Declaración se enumeran varias opciones contenidas en dicha flexibilidad en relación con las licencias obligatorias y el agotamiento. La ulterior decisión de 30 de agosto de 2003, sobre la aplicación del párrafo 6 de la Declaración de Doha (Decisión de 2003) confirma nuevamente los "derechos, obligaciones y flexibilidades que corresponden a los Miembros en virtud de las disposiciones del Acuerdo sobre los ADPIC".61
Sobre la base del Acuerdo entre la Organización Mundial de la Propiedad Intelectual y la Organización Mundial del Comercio, alcanzado el 22 de diciembre de 1995,62 la OMPI proporciona asistencia técnico-jurídica en relación con el Acuerdo sobre los ADPIC. Los organismos gubernamentales encargados de elaborar las leyes suelen solicitar asesoramiento a la OMPI sobre el modo de aplicar en sus países las flexibilidades contenidas en el Acuerdo. Antes de prestar asesoramiento se estudian cuidadosamente las medidas de flexibilidad, su coherencia con el Acuerdo, y sus implicaciones legales, técnicas y económicas. Sin embargo, la decisión definitiva sobre la opción legislativa que se elija compete exclusivamente a cada Estado miembro. Se han determinado cuatro bloques de flexibilidad en la labor de la OMPI:
- el método para aplicar las obligaciones en virtud del Acuerdo sobre los ADPIC;
- normas sustantivas de protección;
- mecanismos de aplicación;
- áreas no contempladas en el Acuerdo.
El uso de los elementos de flexibilidad se aborda también en varias recomendaciones incluidas en la Agenda de la OMPI para el Desarrollo (véase el recuadro 2.9). En respuesta a la petición del Comité sobre Desarrollo y Propiedad Intelectual (CDIP), la OMPI elaboró un estudio preliminar sobre las flexibilidades en materia de patentes en el marco jurídico multilateral y su aplicación legislativa en los planos nacional y regional.63 El estudio presenta un número no exhaustivo de medidas de flexibilidad en la esfera de las patentes, junto con su desarrollo conceptual y varios anexos y cuadros que reflejan las correspondientes disposiciones y prácticas legales de un importante número de países.
El informe mostró una variedad de enfoques para aplicar las flexibilidades previstas en el Acuerdo en las legislaciones nacionales, tales como las licencias obligatorias, las exenciones por investigación, el agotamiento de los derechos, así como la exención basada en el examen reglamentario, también llamada exención Bolar.64 Otro documento amplía esa investigación a otras medidas de flexibilidad, a saber: los períodos de transición, la patentabilidad de sustancias existentes en la naturaleza, las flexibilidades relativas a la divulgación, aspectos relacionados con el examen de fondo y la verificación de oficio por parte de las oficinas de propiedad intelectual de cláusulas anticompetitivas en los acuerdos de concesión de patentes (véase el recuadro 2.10).65
(ii) Antecedentes de la Declaración de Doha
Los negociadores del Acuerdo sobre los ADPIC aspiraban a lograr que los países otorgaran patentes a productos farmacéuticos, pero manteniendo ciertas opciones sobre patentabilidad y alcance de los derechos en interés de la salud pública. Sin embargo, surgió una gran controversia acerca del grado en que el Acuerdo favorecía la salud pública, sobre todo en la época en que entraron en vigor, para los países en desarrollo, la mayoría de las obligaciones sustantivas, en el año 2000.
En una actuación judicial que sentó precedente, una asociación del sector farmacéutico y 39 de sus asociados presentaron denuncias ante el Tribunal Supremo de Pretoria, alegando, entre otras cuestiones, que la legislación de Sudáfrica sobre medicamentos autorizaba la importación paralela de medicamentos (contra la infección por el VIH/sida) y era incompatible con el Acuerdo sobre los ADPIC. La demanda judicial desencadenó una viva campaña, liderada por organizaciones no gubernamentales y militantes contra el sida. En el procedimiento judicial se puso de manifiesto que la legislación de Sudáfrica se había basado en una ley tipo de la OMPI y, al final, en 2001 las empresas retiraron sus denuncias sin condiciones. Para entonces, muchos gobiernos y otros agentes estaban convencidos de que era necesario aclarar la relación entre el Acuerdo sobre los ADPIC y la salud pública.
Recuadro 2.9. Definición de las flexibilidades según la OMPI |
|
These Las flexibilidades se pueden clasificar de diferentes maneras, por ejemplo, agrupándolas con arreglo a la fase del ciclo de vida del derecho de la propiedad intelectual en cuestión. Así pues, las flexibilidades se pueden emplear en relación con: |
|
Recuadro 2.10. Flexibilidades del Acuerdo sobre los ADPIC destacadas en la EMPA-SIP |
La Estrategia mundial y plan de acción sobre salud pública, innovación y propiedad intelectual (EMPA-SIP) de la OMS hace referencia explícita a las flexibilidades que la Declaración de Doha confirma. En ella se insta a los Estados miembros a estudiar la aplicación de las flexibilidades previstas en el Acuerdo sobre los ADPIC, incluidas las reconocidas en la Declaración de Doha, mediante su incorporación a la legislación nacional (elemento 5.2a). En lo que respecta a una protección de la propiedad intelectual que trascienda lo establecido en el Acuerdo, se insta a los Estados miembros a que tengan en cuenta las consecuencias de salud pública al considerar la adopción o aplicación de dichas obligaciones (elemento 5.2b). Los Estados miembros también deberían tener en cuenta las flexibilidades al negociar otros acuerdos comerciales (bilaterales o regionales) (elemento 5.2c). Por otra parte, la EMPA-SIP destaca varias flexibilidades y opciones de política pública a las que pueden recurrir los Estados miembros, concebidas para facilitar la investigación y el acceso a las tecnologías médicas, a saber: l |
|
En abril de 2001, las Secretarías de la OMS y de la OMC convocaron en Høsbjør (Noruega) un taller sobre la fijación diferenciada de precios y financiamiento de medicamentos esenciales. A raíz de la publicación del informe de ese taller72 el Grupo Africano propuso que la OMC convocara una reunión extraordinaria del Consejo de los ADPIC a fin de iniciar los debates sobre la interpretación y aplicación de las disposiciones pertinentes del Acuerdo sobre los ADPIC con miras a aclarar las flexibilidades a las que los Miembros tienen derecho y, en particular, a establecer la relación entre los derechos de propiedad intelectual y el acceso a los medicamentos. Todos los Miembros apoyaron la propuesta.73 Posteriormente, en junio de 2001 un grupo de países en desarrollo elaboró una propuesta detallada por escrito para pedir a la OMC que adoptara medidas para velar por que el Acuerdo no menoscabara de ninguna manera el legítimo derecho de los Miembros de la OMC a formular sus propias políticas de salud pública y ponerlas en práctica adoptando medidas para protegerla. En la Cuarta Conferencia Ministerial de la OMC, celebrada en Doha (Qatar) el 14 de noviembre de 2001, los ministros aprobaron por consenso la Declaración de Doha, en la que se abordaban las preocupaciones que se habían planteado.
(iii) Contenido de la Declaración de Doha
La Declaración integra la función general del Acuerdo sobre los ADPIC en la mejora del acceso a los medicamentos, y aclara las flexibilidades concretas a tal efecto. Así pues, proporciona un marco más claro para adoptar decisiones operativas específicas que permitan aprovechar las opciones de políticas que contempla el Acuerdo.
En la Declaración se reconoce la gravedad de los problemas de salud pública que afligen a muchos países en desarrollo y menos adelantados, especialmente los resultantes de la infección por el VIH/sida, la tuberculosis, el paludismo y otras epidemias. Tras esta aseveración decisiva hubo varias aclaraciones importantes que hacían saber a los Miembros que eran libres de utilizar las disposiciones del Acuerdo de manera que apoyaran los objetivos de salud pública. En el párrafo 4 se confirmaba que "el Acuerdo sobre los ADPIC no impide ni deberá impedir que los Miembros adopten medidas para proteger la salud pública", que "puede y deberá ser interpretado y aplicado de una manera que apoye el derecho de los Miembros de la OMC de proteger la salud pública y, en particular, de promover el acceso a los medicamentos para todos", y, además, que los Miembros tienen el derecho de "utilizar, al máximo, las disposiciones del Acuerdo, que prevén flexibilidad a este efecto".
En el párrafo 5 de la Declaración se confirman concretamente cuatro aspectos en los que las disposiciones del Acuerdo proporcionan flexibilidad a tal efecto:
- La primera aclaración se refiere al modo en que se interpreta el Acuerdo. Cada disposición se leerá a la luz del objeto y fin del Acuerdo tal como se expresa, en particular, en sus "objetivos" y "principios". En la Declaración no se definen esos términos, pero hay un paralelismo con los títulos de los artículos 7 y 8 del Acuerdo, aunque también se hace referencia a objetivos y principios en otros apartados de este.
- Las aclaraciones segunda y tercera se refieren a las licencias obligatorias. Cada Miembro de la OMC tiene "el derecho de conceder licencias obligatorias y la libertad de determinar las bases sobre las cuales se conceden". Así se disipó la idea errónea de que solo se podía recurrir a esas licencias en situaciones de emergencia nacional. Cada Miembro de la OMC tiene además el derecho de determinar lo que constituye una emergencia nacional u otras circunstancias de extrema urgencia. Esas aclaraciones tienen una importancia práctica, ya que en situaciones de ese tipo los países están exentos de negociar previamente una licencia voluntaria con el titular de la patente. En cuanto a ejemplos sobre lo que se puede incluir en ese tipo de emergencias, la Declaración cita "las crisis de salud pública, incluidas las relacionadas con el VIH/sida, la tuberculosis, el paludismo y otras epidemias".
- Por último, en la Declaración se confirma asimismo la libertad de cada Miembro para "establecer su propio régimen para tal agotamiento sin impugnación", a reserva de las normas contra la discriminación en función de la nacionalidad. Así pues, los Miembros pueden elegir entre el agotamiento nacional, regional o internacional.74 El agotamiento determina el grado en que el titular de un derecho de propiedad intelectual puede evitar la reventa y la importación de las mercancías genuinas colocadas en el mercado con su consentimiento en ese país o en algún otro. Por lo tanto, los países pueden decidir libremente si permiten o no la importación paralela de productos patentados, en particular productos médicos.
El párrafo 6 de la Declaración de Doha impulsó el inicio de los trabajos que más tarde culminarían con la aprobación de una flexibilidad adicional, ideada con el objetivo de ayudar a los países cuyas capacidades de fabricación en el sector farmacéutico eran insuficientes o inexistentes a hacer un uso efectivo de las licencias obligatorias.75
El párrafo 7 de la Declaración reafirmó el compromiso de los países desarrollados Miembros de la OMC de ofrecer a sus empresas e instituciones incentivos destinados a fomentar y propiciar la transferencia de tecnología a los Miembros que son países menos adelantados, de conformidad con el párrafo 2 del artículo 66 del Acuerdo; de esa manera, confirmaba que la transferencia de tecnología a los países menos adelantados es también una cuestión de salud pública. Por otra parte, el párrafo 7 encomendaba al Consejo de los ADPIC que ampliara el período de transición para los países menos adelantados, en relación con sus obligaciones en materia de patentes y de protección de datos de pruebas de productos farmacéuticos (incluidos los procedimientos de ejecución y las vías de recurso), hasta el 1º de enero de 2016.
(iv) Aplicación de la Declaración de Doha
A diferencia del Acuerdo sobre los ADPIC, la Declaración de Doha no obliga a adoptar ninguna disposición legislativa concreta. A pesar de ello, algunos países han hecho referencia a su contenido en alguna medida jurídica. Asimismo, se ha mencionado la Declaración en la labor de otras organizaciones internacionales, sobre todo en la EMPA-SIP y muchas otras resoluciones de la OMS, en la Agenda de la OMPI para el Desarrollo, y también en las resoluciones 65/1 y 65/27776 de la Asamblea General de las Naciones Unidas, en las que se abordan los Objetivos de Desarrollo del Milenio y el VIH/sida, respectivamente.
(v) Período de transición para los países menos adelantados
El Acuerdo sobre los ADPIC prevé varios períodos de transición para que los países puedan ejecutar de manera escalonada las obligaciones contraídas. Algunos de esos períodos se centran específicamente en las patentes de los productos farmacéuticos. Si bien los períodos de transición previstos para los países desarrollados y en desarrollo Miembros de la OMC ya han expirado, los países menos adelantados pueden, sobre la base de la Declaración de Doha y la ulterior Decisión del Consejo sobre los ADPIC, beneficiarse de una prórroga del período de transición hasta el 1º de enero de 2016, con respecto a las patentes de productos farmacéuticos y a la protección de datos de pruebas para estos (incluidos los procedimientos de ejecución y las vías de recurso).77 Asimismo, el Consejo General de la OMC aprobó para los países menos adelantados una exención de la obligación dimanante del párrafo 9 del artículo 70 del Acuerdo sobre los ADPIC, lo que también amplió el período de transición hasta el 1º de enero 2016.78 En consecuencia, esos países no están obligados a conceder derechos exclusivos de comercialización de productos farmacéuticos mientras haya solicitudes de patente en trámite, ni siquiera en el caso de los productos que de otro modo entrarían en las circunstancias, muy específicas, que se establecen en el párrafo 9 del artículo 70. Estas decisiones son independientes de la prórroga general del período de transición para esos países con respecto a la mayoría de sus otras obligaciones en el marco de los ADPIC, hasta el 1º de julio 2013.79 Los Miembros que son países menos adelantados pueden obtener otras prórrogas del período de transición presentando una petición debidamente motivada. A este respecto, los ministros presentes en la Octava Conferencia Ministerial de la OMC, celebrada en diciembre de 2011, invitaron al Consejo de los ADPIC a tomar plenamente en consideración una petición debidamente motivada de los Miembros que son países menos adelantados para que se prorrogara su período de transición.80 En noviembre de 2012, el Grupo de Países Menos Adelantados presentó una solicitud para una nueva prórroga del período de transición. Según la propuesta de proyecto de decisión, los países menos adelantados estarían exentos de la aplicación del Acuerdo mientras durase su condición de tales.81 Hasta el momento de redactarse el presente documento, la OMC no había adoptado ninguna decisión al respecto.
En el plano nacional, por lo tanto, los países menos adelantados pueden, por el momento, mantener sus normas jurídicas de protección y observancia sin tener que cumplir con las obligaciones en materia de patentes y de protección de datos de pruebas que se especifican en el Acuerdo con respecto a los productos farmacéuticos. Sin embargo, si esos países desearan rebajar sus estándares de protección mediante patente para los productos farmacéuticos, lo cual sería lícito en virtud de la decisión de prórroga descrita más arriba, por lo general aún tendrían que adoptar las medidas pertinentes para incorporar esos cambios a las leyes nacionales. Así sucedió en Rwanda en 2009, cuando se aprobó una nueva ley sobre la protección de la propiedad intelectual, según la cual no se pueden patentar los productos farmacéuticos a los efectos de los convenios internacionales de los que es signatario ese país.82 En virtud de la anterior legislación sobre patentes de Rwanda, los productos farmacéuticos eran materia patentable. Los países menos adelantados pueden optar por mantener sin cambios la legislación, y simplemente declarar que hasta el final del período de transición no aplicarán las disposiciones legales relativas a la protección de datos de pruebas o a las patentes en la esfera de los productos farmacéuticos. Para cualquiera de esas medidas, esos países tendrán que verificar también la conformidad de la medida prevista con su propio sistema jurídico y con las obligaciones legales que se derivan de su pertenencia a organizaciones regionales o de los acuerdos comerciales bilaterales o de otro tipo de tratados de los que sean signatarios.
Gracias al período de transición, esos países pueden atraer inversiones destinadas a la producción local de productos farmacéuticos. Si83 bien algunos excluyen los productos farmacéuticos de la protección mediante patentes durante el período de transición, otros, como los países menos adelantados miembros de la Organización Africana de la Propiedad Intelectual, han renunciado a esa opción debido a que el Acuerdo de Bangui prevé la concesión de patentes farmacéuticas.84
(h) Condiciones de adhesión a la OMC
Estas condiciones son otra posible fuente de obligaciones en materia de propiedad intelectual en el sistema de la OMC. Los aspirantes a ser Miembros tienen que negociar su adhesión a la OMC de conformidad con el artículo XII del Acuerdo por el que se establece la Organización Mundial del Comercio.85 Las condiciones son, por lo tanto, negociables. Las negociaciones se producen entre el miembro en proceso de adhesión y los Miembros ya pertenecientes a la OMC que tengan interés y se presten a participar en el Grupo de Trabajo sobre la Adhesión. Como mínimo, las condiciones estipulan siempre el cumplimiento de todos los Acuerdos multilaterales de la OMC, incluido el Acuerdo sobre los ADPIC, a reserva de posibles períodos de transición. En varias ocasiones, los Miembros ya pertenecientes a la OMC han solicitado además el cumplimiento de ciertos compromisos adicionales. Si el miembro en proceso de adhesión acepta cumplirlos, esos compromisos se indican en el informe del Grupo de Trabajo y se citan en el Protocolo de Adhesión, que forma parte del Acuerdo sobre la OMC para ese Miembro. Puede ocurrir que los nuevos Miembros acepten condiciones de adhesión por las que se exigen mayores niveles de protección en materia de propiedad intelectual que los que se prevén en el Acuerdo sobre los ADPIC. Sin embargo, no todos los elementos del informe del Grupo de Trabajo tienen el mismo valor jurídico. Algunos de ellos son compromisos jurídicamente vinculantes, y se detallan en el informe y en el Protocolo de Adhesión; pero hay otros elementos cuya naturaleza es simplemente descriptiva, y reflejan simplemente la información que el país en proceso de adhesión ha facilitado al Grupo de Trabajo. En tal caso, este último no hace constar obligación alguna con respecto a esos elementos.
Han surgido cuestiones relativas a la propiedad intelectual y a los productos farmacéuticos en varias negociaciones de adhesión (para conocer una descripción completa de los elementos de propiedad intelectual en los acuerdos de adhesión a la OMC, véase Abbott y Correa (2007)). Por ejemplo, cuando Ucrania se adhirió a la OMC en 2008, registró el compromiso de informar a los primeros solicitantes de autorización de comercialización de productos farmacéuticos originarios cuando se presentaran solicitudes ulteriores. El objetivo era dar a los primeros solicitantes la posibilidad de presentar información sobre si los solicitantes ulteriores tenían o no permiso para usar los datos originales de las pruebas, y conceder derechos exclusivos sobre esos datos durante al menos cinco años.86
Recuadro 2.11. Condiciones de adhesión a la OMC para un país menos adelantado: el ejemplo de Camboya |
Camboya fue el primer país menos adelantado en concluir las negociaciones de adhesión a la OMC (muchos PMA eran Miembros originales de la OMC cuando esta se creó en 1995). El Grupo de Trabajo sobre la adhesión de Camboya se formó en 1994, y celebró reuniones de 2001 a 2003. Camboya se adhirió a la OMC en 2004. En sus condiciones de adhesión, ese país se comprometió a aplicar el Acuerdo sobre los ADPIC a más tardar el 1º de enero de 2007, a pesar de que en la Declaración de Doha se había acordado conceder a los Miembros que son PMA una prórroga hasta el 1º de enero de 2016 con respecto a las patentes y a la protección de datos de pruebas correspondientes a productos farmacéuticos, y de que posteriormente se acordó concederles una prórroga general hasta el 1º de julio de 2013. |
El compromiso de Camboya de aplicar el Acuerdo a partir de 2007 se hizo en el entendimiento de que durante el período de transición, entre otras cosas, concedería derechos exclusivos para los datos de pruebas durante cinco años y estipularía la vinculación de patentes en las autorizaciones de comercialización (documento de la OMC WT/ACC/KHM/21, párrafos 204-206 y 224). Así pues, Camboya aceptó exigencias de Miembros de la OMC que iban más allá de las obligaciones expresamente establecidas en el Acuerdo. Todo indicaba que Camboya, en su acuerdo de adhesión, había renunciado a varias de las flexibilidades previstas en el Acuerdo, de las que de otro modo podría haberse beneficiado, en virtud de los períodos de transición en curso. |
Sin embargo, en el período inmediatamente anterior a la adopción de la decisión sobre la adhesión de Camboya, el Director General Adjunto de la OMC, en nombre del Presidente del Grupo de Trabajo sobre la Adhesión de Camboya, aclaró que: "Los resultados alcanzados en el caso de Camboya hablan por sí solos y, en ese contexto, también debo añadir que sus condiciones de adhesión no excluyen que Camboya, en su calidad de país menos adelantado, pueda acogerse a los beneficios previstos en la Declaración de Doha relativa al Acuerdo sobre los ADPIC y la Salud Pública" (documento de la OMC WT/MIN(03)/SR/4). |
Con respecto a los países menos adelantados (PMA), en la Declaración Ministerial de 2001, que puso en marcha el Programa de Doha para el Desarrollo, se acordó que los Miembros de la OMC se esforzarían por facilitar y acelerar las negociaciones con los PMA en proceso de adhesión. En 2002, el Consejo General de la OMC adoptó las directrices sobre la adhesión de los PMA.87 En las directrices se prevé, entre otras cuestiones, que se establecerán los períodos de transición previstos en determinados Acuerdos de la OMC -teniendo en cuenta las necesidades individuales en materia de desarrollo, finanzas y comercio- y que esos períodos irán acompañados de Planes de Acción para el cumplimiento de las normas de comercio. Además, en una decisión adoptada en la Octava Conferencia Ministerial de la OMC, en diciembre de 2011, se estipuló que "las solicitudes de períodos de transición adicionales se examinarán teniendo en cuenta las necesidades individuales en materia de desarrollo de los PMA en proceso de adhesión".88 Posteriormente, la Decisión del Consejo General de la OMC de 25 de julio de 2012 hizo más simples y operativas las directrices sobre la adhesión de los PMA, entre otras cosas mediante una mayor transparencia y el compromiso de que los períodos de transición adicionales se considerarían favorablemente, caso por caso.89 Camboya y Nepal se adhirieron a la OMC en 2004, Cabo Verde en 2008, y Samoa y Vanuatu en 2012 (véase el recuadro 2.11).
2. Política de competencia
Entre los instrumentos normativos de que disponen los gobiernos para hacer frente a los problemas de salud pública, la política de competencia es un factor importante para lograr el acceso a la tecnología médica y fomentar la innovación en el sector farmacéutico. La competencia propicia la libertad de elección, la disminución de los precios y la buena relación calidad-precio, e impulsa notablemente la innovación y la productividad.
(a) La doble función de la política de competencia
Al examinar las políticas destinadas a fomentar la innovación y mejorar el acceso a las tecnologías médicas, puede considerarse que la política de competencia tiene dos funciones interrelacionadas que se complementan entre sí (Hawkins, 2011).
En primer lugar, dicha política es importante para adoptar con conocimiento de causa medidas reglamentarias y otras decisiones normativas pertinentes en relación con la innovación de las tecnologías médicas y el acceso a ellas. Los órganos supervisores de la competencia pueden tener como mandato la realización de exámenes generales de las políticas de competencia y reglamentación, los regímenes de regulación de precios farmacéuticos, la reglamentación de las farmacias y los acuerdos de venta o distribución al por mayor. Pueden formular recomendaciones normativas en relación con diversas políticas que afectan a la competencia, no solo en lo que respecta al funcionamiento de las leyes de competencia y de protección del consumidor, sino también en áreas relacionadas directamente con la salud pública. Instituciones como la Organización de Cooperación y Desarrollo Económicos y el Banco Mundial han publicado estudios sobre la interrelación entre la política de competencia y la reglamentación sanitaria. Dicha interrelación fomenta la coordinación entre las autoridades supervisoras de la competencia y los organismos que regulan los precios de los productos médicos y el sector sanitario en general.90
En segundo lugar, la aplicación de la legislación sobre competencia ayuda asimismo a corregir conductas anticompetitivas que puedan producirse en los diversos sectores de actividad económica que intervienen en el desarrollo de tecnologías médicas y en el suministro a los pacientes que las necesitan. Apunta también a prevenir prácticas anticompetitivas que pueden, por ejemplo, restringir la investigación y el desarrollo; limitar la disponibilidad de los recursos necesarios para producir tecnologías médicas; crear obstáculos innecesarios a la introducción de los productos genéricos en el mercado y a la competencia entre marcas; y, en general, limitar los canales de distribución y las posibilidades de elección del consumidor. Entre las prácticas que se han reconocido como perjudiciales en ese sentido se incluyen las siguientes (pero no se limitan a ellas): i) abusos de los derechos de propiedad intelectual debidos a la negativa de negociar condiciones excesivamente restrictivas o a la imposición de ese tipo de condiciones en la concesión de licencias de tecnologías médicas; ii) impedir la competencia de los productos genéricos mediante acuerdos sobre patentes de carácter anticompetitivo; iii) fusiones entre empresas farmacéuticas que conducen a una concentración desaconsejable de actividades de investigación y desarrollo y de derechos de propiedad intelectual; iv) acuerdos de creación de cárteles entre empresas farmacéuticas, en particular entre fabricantes de productos genéricos; v) conducta anticompetitiva en el sector de la venta al por menor de productos médicos y otros sectores conexos; y vi) manipulación de las licitaciones en la contratación pública. Esas prácticas se pueden abordar, caso por caso, basándose en la aplicación de la legislación sobre competencia.
(b) La interrelación entre la política de competencia y la protección de la propiedad intelectual
En la esfera de la innovación, los objetivos y efectos de la protección de la propiedad intelectual y la política de competencia pueden ser complementarios: ambos están orientados a fomentar la innovación creando incentivos para la obtención de nuevos productos como ventaja frente a los competidores. La protección de la propiedad intelectual de las tecnologías médicas novedosas suele considerarse un medio importante para fomentar la inversión en investigación y desarrollo en esta esfera. Favorece la competencia entre empresas fabricantes de productos originarios en lo que respecta al desarrollo de nuevas tecnologías médicas valiosas, y, por ende, acelera su producción y disponibilidad. Por lo general, los derechos de propiedad intelectual no suelen obstaculizar esa forma de competencia, sino que la refuerzan. La política de competencia ayuda asimismo a mantener el potencial innovador de la industria regulando la estructura de mercado y proporcionando contramedidas para las conductas anticompetitivas. Las autoridades supervisoras de la competencia fiscalizan las fusiones de las empresas farmacéuticas y pueden someter a venta o cesión determinadas ramas de investigación a fin de evitar que se abandone la investigación de tecnologías médicas que en un futuro podrían ser competitivas.91 En condiciones ideales, esto debería conducir a lo que se conoce como competencia entre patentes en el mercado farmacéutico: es decir, habría en el mercado varios productos de la misma categoría terapéutica y en ese caso los fabricantes competirían en el mismo mercado.
Sin embargo, a pesar de que mediante los derechos de propiedad intelectual se pretende estimular la innovación, en determinadas circunstancias pueden impedir o disminuir la competencia en el sector farmacéutico en la fase de fabricación, ya que los competidores no pueden utilizar la tecnología médica patentada o sujeta a protección. En ese sentido, es importante considerar el grado en que se dispone de otros productos similares. Cuando hay competencia de otros productos, los derechos de propiedad intelectual no conducen a la creación de monopolios económicos.
En consecuencia, los encargados de la formulación de políticas se enfrentan a la difícil tarea de encontrar un equilibrio global entre la protección y la observancia de los legítimos derechos de propiedad intelectual y la necesidad de estimular la competencia y evitar conductas anticompetitivas.
(i) Cómo abordar las inquietudes relativas a la política de competencia en el marco jurídico para la protección de la propiedad intelectual
La política de competencia ha sentado las bases del marco jurídico para la protección de la propiedad intelectual, puesto que los acuerdos internacionales y las leyes nacionales sobre el particular le otorgan una importante función de "freno y contrapeso" en este ámbito. Las disposiciones jurídicas sobre competencia pueden considerarse parte esencial de las normas sobre protección de la propiedad intelectual.
En el plano internacional, el Convenio de París reconoció hace tiempo la pertinencia de la política de competencia en la concepción de las normas de protección de la propiedad intelectual como base para la concesión de licencias obligatorias destinadas a prevenir el abuso de los derechos de propiedad intelectual. Su pertinencia se refleja asimismo en varias disposiciones del Acuerdo sobre los ADPIC.
El párrafo 2 del artículo 8 del Acuerdo estipula que podrá ser necesario aplicar medidas apropiadas, siempre que sean compatibles con lo dispuesto en el Acuerdo, para prevenir el abuso de los derechos de propiedad intelectual por sus titulares o el recurso a prácticas que limiten de manera injustificable el comercio o redunden en detrimento de la transferencia internacional de tecnología. Como puede verse, esa disposición no se limita necesariamente a las violaciones de la legislación sobre la competencia, sino que abarca el concepto, sin duda más general, de "abuso" de los derechos en cuestión.
En una esfera afín, pero centrándose en el tema concreto de las prácticas relativas a la concesión de las licencias que restringen la competencia, el párrafo 1 del artículo 40 del Acuerdo recoge la convicción de los Miembros de la OMC de que ciertas prácticas o condiciones relativas a la concesión de las licencias de los derechos de propiedad intelectual, que restringen la competencia, pueden tener efectos perjudiciales para el comercio y pueden impedir la transferencia y la divulgación de tecnologías novedosas. Para hacer frente a esta inquietud, en el párrafo 2 del artículo 40 del Acuerdo se reconoce el derecho de los gobiernos Miembros de la OMC a adoptar medidas para impedir abusos de los derechos de propiedad intelectual que tengan un efecto negativo en la competencia. En el mismo párrafo se ofrecen algunos ejemplos de prácticas que pueden considerarse abusivas. Se trata de condiciones exclusivas de retrocesión, condiciones que impidan la impugnación de la validez y licencias conjuntas obligatorias.92
En virtud del artículo 31 del Acuerdo, por el que se establecen determinadas condiciones para utilizar una patente sin autorización del titular de los derechos, el apartado k) deja claro que los Miembros no están obligados a aplicar algunas de esas condiciones en situaciones en las que la licencia obligatoria se concede "para poner remedio a prácticas que, a resultas de un proceso judicial o administrativo, se haya determinado que son anticompetitivas". Es decir, se debe mostrar que el usuario potencial ha intentado obtener la autorización del titular de los derechos en términos y condiciones comerciales razonables y que esos intentos no han surtido efecto en un plazo prudencial, y además que la autorización para usar una patente en virtud de una licencia obligatoria será principalmente para abastecer el mercado interno del Miembro que autorice tales usos. Por otra parte, las autoridades pueden tener en cuenta la necesidad de corregir las prácticas anticompetitivas al determinar el monto de la remuneración debida.
En muchos países, la legislación nacional sobre propiedad intelectual por la que se aplica el Acuerdo también reconoce la importancia de la política de competencia en el ámbito de los derechos de dicha propiedad. Por ejemplo, la Ley de Patentes de la India prevé la concesión de licencias obligatorias sin la condición de haber intentado previamente obtener una autorización del titular de la patente, en términos y condiciones razonables, en el caso de que este haya adoptado prácticas anticompetitivas (apartado iv) de la sección 84.6), así como el derecho a exportar cualquier producto fabricado con arreglo a dichas licencias, si fuese necesario.
(ii) Aplicación de la legislación sobre competencia en el contexto de la propiedad intelectual
La legislación específica es un instrumento útil para corregir, caso por caso, los abusos en materia de derechos de propiedad intelectual.93 En términos generales, no hay ningún principio especial de la legislación sobre competencia que se aplique a la propiedad intelectual, y la protección de esta no exime de aplicar las disciplinas de la legislación sobre competencia. Tampoco cabe presumir que la protección de dicha propiedad confiere poder de mercado o indica una conducta anticompetitiva. De hecho, se considera que los derechos de propiedad intelectual ayudan a crear mercados eficientes y fomentar la innovación. La legislación sobre competencia no impide, como norma general, que los titulares de los derechos de propiedad intelectual ejerzan sus derechos exclusivos. Este respeto global de los derechos de propiedad intelectual en virtud de la legislación sobre competencia se basa en el supuesto de que esos derechos se adquirieron legítimamente mediante un sistema que no confiere derechos excesivamente amplios.
Por tanto, la función de la legislación sobre competencia es proporcionar medidas "correctoras" solo cuando sea necesario. La aplicación de medidas coercitivas en virtud de dicha legislación puede estar justificada cuando el sistema de protección de la propiedad intelectual no pueda impedir por sí mismo las restricciones de la competencia no deseadas.
3. Pautas de política comercial
Todos los países dependen en mayor o menor grado de los productos importados para atender las necesidades de atención sanitaria de su población; en la mayor parte, especialmente los países en desarrollo más pequeños cuya capacidad de producción local de tecnologías médicas es baja o nula, los productos importados contribuyen de manera decisiva a sostener el sistema nacional de salud. Además, los países participan cada vez más en el comercio de servicios de atención sanitaria. Por lo tanto, la política comercial repercute en el modo en que los mercados de tecnologías médicas se abren a la competencia de bienes y servicios importados.
Las normas de comercio internacional se establecen en el ámbito multilateral en el marco de la OMC. Una de las piedras angulares de la Organización es el principio de no discriminación en las relaciones comerciales internacionales, que se pone en práctica mediante los principios de trato nacional y trato de la nación más favorecida. Estos principios están consagrados en el Acuerdo General sobre Aranceles Aduaneros y Comercio (GATT) en relación con el comercio de bienes, en el Acuerdo General sobre el Comercio de Servicios (AGCS) y en el Acuerdo sobre los ADPIC en relación con la propiedad intelectual. En el caso del GATT y el AGCS se aplican importantes excepciones, en particular el trato especial y diferenciado en favor de los países en desarrollo y los acuerdos de libre comercio.
La OMC también garantiza a sus Miembros el derecho de proteger la salud pública. Desde sus comienzos en 1947, el GATT ha otorgado a los países el derecho a tomar las medidas de restricción del comercio que sean necesarias para proteger la salud y la vida de las personas y de los animales o para preservar los vegetales en determinadas condiciones establecidas en el apartado b) del artículo XX. El AGCS contiene una excepción similar en lo que respecta al comercio de servicios, en el apartado b) de su artículo XIV. Esas excepciones generales pueden invalidar las obligaciones y los compromisos adoptados en el marco de la OMC, siempre y cuando las medidas sanitarias, y la forma en que se aplican, cumplan ciertas condiciones. Además, el artículo 8 del Acuerdo sobre los ADPIC reconoce el derecho de los Miembros a adoptar las medidas necesarias para proteger la salud pública, siempre que esas medidas sean compatibles con lo dispuesto en el Acuerdo.
(a) Aranceles
Los aranceles o derechos de aduana que se aplican a las mercancías importadas son un instrumento clásico de política comercial y, en el marco de las normas de la OMC, se prefieren a las restricciones cuantitativas tales como los contingentes, que están prohibidos en general. Los aranceles son relativamente transparentes y, a diferencia de los contingentes, no imponen restricciones estrictas sobre el volumen de las importaciones.
Los Miembros de la OMC han acordado determinados niveles máximos de los aranceles que aplican a todos o a la mayoría de los productos importados, incluidos los farmacéuticos. Esos niveles máximos se llaman "consolidaciones arancelarias" y varían en función del país y del producto. Son el resultado de decenios de negociaciones arancelarias que han permitido obtener consolidaciones para un número cada vez mayor de productos, lo cual crea un entorno comercial más previsible y estable. Las rondas sucesivas de negociaciones han permitido también bajar los tipos arancelarios consolidados y, de hecho, los Miembros de la OMC suelen aplicar aranceles inferiores al tipo consolidado. Por ejemplo, los países en desarrollo han consolidado sus aranceles sobre formulaciones en un promedio de 22,4% ad valorem (calculado sobre el valor de las importaciones), pero, en realidad, aplican en promedio un arancel de 3,4% ad valorem.94
A causa de los aranceles los productos importados, incluidos los medicamentos, resultan más caros para los consumidores. No obstante, muchos países aplican aranceles para favorecer la posición competitiva de las empresas locales en el mercado nacional con el fin de preservar el empleo o promover el desarrollo de la rama de producción (por ejemplo, la capacidad de producción local del sector farmacéutico) o mantener cierto nivel de independencia con respecto a los mercados internacionales. Para los consumidores, la protección arancelaria puede tener efectos encarecedores. Además, los aranceles aumentan los ingresos de los gobiernos, aunque, en el caso de los medicamentos, la cuantía de esos ingresos no suele ser significativa.
En los países desarrollados, los aranceles aplicados a los medicamentos son muy bajos o incluso inexistentes. Varios Miembros de la OMC, países desarrollados principalmente, alcanzaron en 1994 el Acuerdo para la eliminación de aranceles sobre productos farmacéuticos, por el cual eliminaron los aranceles sobre todos los productos farmacéuticos acabados, así como sobre determinados principios activos e insumos para manufactura. Desde 1994, las Partes han actualizado periódicamente la cobertura del acuerdo. Desde 2000, los países desarrollados han aplicado sobre los medicamentos aranceles inferiores al 0,1% ad valorem, en promedio. En el caso de los países en desarrollo, en el último decenio han bajado los tipos arancelarios que aplican a los medicamentos, en promedio, del 6,7% al 4,2%. Entre ellos se incluyen algunos países con industrias manufactureras locales que aplican aranceles relativamente elevados a los productos terminados. En el caso de los países menos adelantados, los tipos arancelarios medios van desde un 4,5% hasta un 2%.
A menudo se conceden exenciones arancelarias sobre ciertos medicamentos o a ciertos compradores. El sector público y los compradores privados sin ánimo de lucro suelen gozar de exenciones arancelarias. Health Action International (HAI), en colaboración con la OMS, ha iniciado un importante proyecto para determinar los diversos costos asociados a los precios de los medicamentos en varios países. En el caso de algunos países, los datos proporcionan información sobre aranceles y exenciones.95
(b) Medidas no arancelarias
Gracias a las rondas sucesivas de negociaciones celebradas en los últimos 60 años se ha logrado una disminución constante de los tipos arancelarios, lo que ha causado un cambio de enfoque hacia otro tipo de medidas comerciales. Algunos expertos sostienen que estas últimas están sustituyendo progresivamente a los aranceles como medida de protección de las industrias nacionales. Son medidas no arancelarias, entre otras: las medidas sanitarias, los reglamentos técnicos, las inspecciones previas a la expedición, las licencias de importación, las medidas de control de precios, las cargas e impuestos, las restricciones a la distribución y los servicios de posventa. Varios Acuerdos de la OMC abordan ese tipo de medidas con el objetivo básico de establecer normas para su uso a fin de que no se conviertan en obstáculos innecesarios al comercio. Si bien todas esas medidas pueden repercutir en el comercio de los productos farmacéuticos, las dos que se describen a continuación tienen consecuencias directas sobre la salud pública.
(i) Medidas sanitarias y fitosanitarias
El Acuerdo de la OMC sobre la Aplicación de Medidas Sanitarias y Fitosanitarias (Acuerdo MSF) contiene normas específicas para los países con el fin de velar por la inocuidad alimentaria y evitar la transmisión a las personas de enfermedades animales o vegetales por medio del comercio. Se aspira a lograr un equilibrio entre el reconocimiento del derecho soberano de los Miembros a determinar el grado de protección sanitaria que consideren apropiado, y evitar que las prescripciones sanitarias o fitosanitarias representen una restricción innecesaria, arbitraria, científicamente injustificable o encubierta al comercio internacional. El Acuerdo MSF estipula que esas medidas no deben entrañar un grado de restricción del comercio mayor del requerido para lograr un nivel adecuado de protección sanitaria o fitosanitaria, teniendo en cuenta su viabilidad técnica y económica. Por lo tanto, alienta a los Miembros a que cumplan con las normas, directrices y recomendaciones internacionales. Los Miembros pueden adoptar medidas sanitarias o fitosanitarias que aumenten el nivel de protección sanitaria, o medidas para las cuales no existan normas internacionales, a condición de que estén basadas en principios científicos.96
(ii) Obstáculos técnicos al comercio
El Acuerdo sobre Obstáculos Técnicos al Comercio (Acuerdo OTC) abarca las prescripciones técnicas de los productos no recogidas en el Acuerdo MSF. Rige tanto las prescripciones de carácter obligatorio (los "reglamentos técnicos") como las de carácter facultativo (las "normas"), además de los procedimientos de evaluación de la conformidad con dichas prescripciones, tales como las inspecciones. Entre los reglamentos técnicos y las normas se incluyen, por ejemplo, las prescripciones de calidad para los productos farmacéuticos, las prescripciones de etiquetado para los productos alimenticios y las normas de seguridad para los aparatos de rayos X. El Acuerdo OTC incorpora el principio de la no discriminación, tanto en el sentido de trato nacional como de nación más favorecida. Asimismo, estipula que los reglamentos técnicos no restringirán el comercio más de lo necesario para alcanzar un objetivo legítimo, teniendo en cuenta los riesgos que crearía no alcanzarlo. La protección de la salud o seguridad humanas figura como objetivo legítimo. En otras palabras, el Acuerdo permite a los países regular el comercio a fin de proteger la salud, pero exige que las medidas que se adopten no supongan una restricción innecesaria del comercio. Asimismo, se alienta a los Miembros a que basen sus medidas en normas internacionales, aunque pueden apartarse de ellas si consideran que su aplicación sería ineficaz o inapropiada para el cumplimiento de determinados objetivos legítimos.97
(c) Comercio de servicios
Los servicios de salud contribuyen muchísimo a lograr la disponibilidad real y al uso adecuado de muchos productos farmacéuticos y de otras tecnologías médicas; cabe mencionar especialmente los servicios que se ocupan de la prevención, el diagnóstico y el tratamiento, pero también los servicios auxiliares y de apoyo técnico. En el caso de muchos servicios de diagnóstico o regímenes terapéuticos avanzados, no hay una distinción clara entre el acceso efectivo y adecuado a la tecnología como tal y la prestación de los servicios conexos. Las decisiones que se adopten en relación con la apertura de los servicios de salud a proveedores extranjeros pueden, por tanto, repercutir en el acceso a las tecnologías médicas.
(i) El marco jurídico multilateral
El AGCS es el principal instrumento jurídico multilateral por el que se rige el comercio de servicios de salud, que se define como la prestación de un servicio mediante cuatro "modos de suministro" diferentes, todos ellos pertinentes para el sector sanitario:
- Modo 1: suministro transfronterizo (por ejemplo, la telemedicina).
- Modo 2: consumo en el extranjero (por ejemplo, un paciente que se desplace a un país extranjero para recibir tratamiento médico).
- Modo 3: establecimiento de una presencia comercial (por ejemplo, una clínica abre una filial en el extranjero, o invierte en unas instalaciones ya existentes situadas en el extranjero).
- Modo 4: presencia de personas físicas (por ejemplo, un médico se desplaza al extranjero para trabajar en una clínica de propiedad extranjera).
(ii) Alcance de los compromisos del AGCS en los sectores relacionados con la salud
El AGCS concede a los Miembros de la OMC completa flexibilidad para decidir qué sectores y modos de suministro abren a la competencia extranjera, así como el nivel de obligaciones que están dispuestos a contraer. Los servicios de salud se dividen en varias categorías: i) servicios de hospital; ii) otros servicios de salud humana; iii) servicios sociales; iv) servicios médicos y odontológicos; y v) servicios prestados por comadronas, enfermeras, fisioterapeutas y personal paramédico.98 Hay otros servicios que complementan y facilitan el acceso a las tecnologías médicas, como por ejemplo: investigación y desarrollo en medicina; farmacia, venta mayorista y minorista de diversos productos farmacéuticos y productos y dispositivos médicos y quirúrgicos; servicios de mantenimiento y reparación de equipos médicos; y servicios de ensayos y análisis técnicos. Las disciplinas del AGCS no abarcan los servicios "suministrados en ejercicio de facultades gubernamentales" (es decir, los que no se suministran ni "en condiciones comerciales" ni "en competencia con uno o varios proveedores de servicios"). Debido a ello, muchos servicios de salud del sector público quedan fuera del ámbito de aplicación del AGCS.
Muchos países han liberalizado progresivamente sus servicios de salud, lo que ha creado más oportunidades para la empresa privada. Sin embargo, siguen siendo reacios a hacer que esa apertura sea vinculante con arreglo al AGCS. Dejando a un lado los servicios de seguro de enfermedad, hay menos compromisos contraídos en el marco del AGCS sobre servicios de salud que sobre cualquier otro sector (véase el cuadro 2.4). Posiblemente ello se deba a la importante función que desempeñan las entidades públicas en la prestación de servicios de salud, junto con la cuestión de las sensibilidades políticas y la ausencia de intereses comerciales manifiestos. Los servicios de salud no han sido objeto de negociaciones bilaterales activas, y los compromisos que determinados países han adoptado en ese sector han surgido principalmente por iniciativa propia (Adlung, 2010). En cualquier caso, es importante señalar que comprometerse a abrir el sector de servicios a la competencia extranjera no afecta a la capacidad de un gobierno para regular el sector.
En los seis sectores sanitarios considerados, en general hay reticencia a introducir obligaciones relativas al suministro transfronterizo de servicios de salud, probablemente porque no se sabe con seguridad cómo formular y hacer cumplir la reglamentación pertinente a proveedores de servicios situados en el extranjero (un comportamiento que también se observa en otros sectores de servicios). Los compromisos relativos a los servicios de salud que se consumen en el extranjero representan la mayor parte de los compromisos plenos, lo que puede ser un reflejo de la reticencia -y la incapacidad- de los gobiernos para impedir que sus ciudadanos salgan al extranjero a consumir servicios (práctica que se da en todos los sectores de servicios). Algunos Miembros limitan la posibilidad de transferir los seguros para que cubran los tratamientos recibidos en el exterior, lo que puede disuadir a los pacientes de solicitar tratamiento en el extranjero. Casi la mitad de los compromisos relacionados con el suministro de servicios de salud mediante presencia comercial se consolidaron sin limitaciones a escala sectorial, un resultado que parece superior a la media de los demás sectores.99 La mayoría de los compromisos en el marco de este modo de suministro están sujetos a limitaciones, por ejemplo, límites a la participación extranjera y prescripciones en materia de empresas conjuntas o de residencia. Algunos consignan pruebas de necesidades económicas; es decir, antes de autorizar la creación de nuevos hospitales o clínicas, se consideran criterios tales como la densidad de población, los centros médicos ya existentes, el grado de especialización, el tipo de material médico, y la distancia o la infraestructura de transporte existente.
A diferencia de los otros modos de suministro, los compromisos en materia de servicios de salud proporcionados mediante la presencia de personas físicas han sido abordados de forma "horizontal" por la gran mayoría de los Miembros, de suerte que se aplican a todos los sectores de servicios abarcados. La mayoría de los Miembros de la OMC han consignado condiciones muy restrictivas en relación con este modo de suministro, reservadas a personas muy calificadas o personas ligadas a una presencia comercial, y no a personas que trabajan por cuenta propia (WTO, 2009). Algunos restringen aún más sus compromisos, e incluyen requisitos relativos a lengua, residencia o nacionalidad, reconocimiento de títulos, estrictos límites de tiempo, pruebas de necesidades económicas o contingentes, con lo que reducen aún más el ya limitado nivel de consolidaciones. Hay indicios, sin embargo, de que en la práctica los profesionales de la salud obtienen mejores condiciones de acceso que las que obtendrían si se vieran limitados exclusivamente a las consolidaciones del AGCS. Los compromisos en materia de servicios de salud también presentan limitaciones relativas a la amplitud de las actividades abarcadas, como las exclusiones de los proveedores públicos, las restricciones de los compromisos sobre los servicios de hospital prestados por el sector privado o con financiación privada, o los tipos de especialización médica abarcados.
(iii) La creciente importancia económica del comercio de servicios de salud y la repercusión de los compromisos del AGCS
Según Gottret y Schieber (2006), la atención sanitaria es probablemente el principal sector de actividad económica del mundo: la cifra combinada de negocios es de más de 3,2 billones de dólares EE.UU. al año, equivalente a la décima parte del producto interno bruto (PIB) mundial, y da empleo a más de 59 millones de personas. Los servicios de salud siguen globalizándose mediante la circulación transfronteriza de trabajadores de la salud y de pacientes, así como mediante las inversiones de las empresas de servicios de salud (OMS/OMC, 2002; Blouin et al., 2006). Los avances tecnológicos y la disminución del costo de las telecomunicaciones han favorecido la aparición de la telemedicina en diversos procedimientos de salud (por ejemplo, la radiología, el diagnóstico, los exámenes anatomopatológicos, las interconsultas y la cirugía a distancia). Es casi imposible medir la repercusión de los compromisos del AGCS en los servicios de salud -y en cualquier otro sector- porque los datos son limitados y es complicado distinguir los efectos de las consolidaciones de políticas comerciales de los efectos de otras medidas normativas y reglamentarias. Sin embargo, los estudios indican que la repercusión de los compromisos del AGCS en la estructura del comercio ha sido probablemente insignificante. Esos compromisos no implican una mayor liberalización, pero (en el mejor de los casos) consolidan los niveles ya existentes de acceso a los mercados. En consecuencia, la comercialización de los servicios de salud se ha producido con independencia de las obligaciones del AGCS, y el efecto principal de ese acuerdo parece haber sido hacer más previsibles las políticas nacionales (Adlung, 2010).
Cuadro 2.4. Número de compromisos del AGCS |
|||||||
|
Servicios médicos y odontológicos |
Enfermeras, comadronas, etc. |
Servicios de hospital |
Otros servicios de salud humana |
Servicios sociales |
Otros |
Servicios de seguro de enfermedad |
Number of commitments |
65 |
35 |
57 |
26 |
27 |
6 |
103 |
Fuente: Secretaría de la OMC (los Estados miembros de la Unión Europea se han contabilizado por separado).
Recuadro 2.12. Código de prácticas mundial de la OMS sobre contratación internacional de personal de salud
|
A fin de mejorar la reglamentación en materia de migración y circulación de personal sanitario en las zonas donde más se lo necesita, la OMS elaboró este Código que tiene estos elementos esenciales: |
|
|
|
|
El sector sanitario ha estado prácticamente ausente de las negociaciones en materia de servicios en el marco de la Ronda de Doha de la OMC; solo alrededor de una docena de Miembros, en su mayoría países en desarrollo, presentaron ofertas relativas a ese sector. Las ofertas fueron en general muy restrictivas (relativas a un único modo o a especializaciones médicas particulares). Otros, tales como el Canadá, Suiza o la Unión Europea, excluyeron explícitamente la salud y otros servicios sociales de las negociaciones en el marco de la OMC. Esa falta generalizada de interés se puede atribuir al papel preponderante del sector público en la prestación de servicios de atención sanitaria, aunado a la importante dimensión de servicio público y social y a la cautela de no limitar las opciones de políticas para el futuro.
(iv) Problemas ligados a la apertura del comercio de servicios de salud
La apertura del comercio de servicios de salud no debe considerarse un fin en sí mismo, sino una herramienta que puede ser claramente beneficiosa si se utiliza correctamente en un contexto normativo más amplio. Desde la perspectiva de la salud pública, intensificar el comercio de servicios puede crear tanto oportunidades para mejorar la prestación de servicios de salud como riesgo de desigualdades, si solo aquellos que pueden permitírselo consiguen acceder a los nuevos servicios de salud transfronterizos. A menudo se expresa la preocupación de que la apertura de los servicios de salud puede crear un sistema de dos niveles -buenos servicios para los ricos, malos servicios para los pobres-, lo que pondría en peligro el acceso equitativo de todos. Por ejemplo, la exportación de servicios de salud desde centros deslocalizados a través de Internet podría aumentar enormemente las oportunidades de empleo en los países en desarrollo, y contener el gasto en los países desarrollados; sin embargo, el personal sanitario se vería atraído por mejores ofertas económicas, lo que podría crear deficiencias en el sector sanitario local.
Es imprescindible que haya un sistema reglamentario sólido y fiable para velar por que la actuación de los proveedores privados contribuya a hacer frente a cuestiones más amplias de política pública, tales como el acceso equitativo y asequible para todos. Los establecimientos sanitarios de propiedad y gestión pública también entrañan problemas de reglamentación. Así pues, es preciso contar con un marco reglamentario apropiado para velar por que la apertura del comercio de servicios de salud beneficie a todos los sectores de la población. Antes de adquirir compromisos vinculantes con arreglo al AGCS o a cualquier otro acuerdo comercial, debería realizarse una evaluación de sus efectos sobre el suministro de dichos servicios. La migración del personal sanitario es una cuestión determinante, ya que los trabajadores tienden a emigrar de las regiones más pobres a ciudades más ricas dentro de un país, y, de allí, a países de ingresos elevados (véase el recuadro 2.12). En esos países la demanda de agentes sanitarios extranjeros ha aumentado debido a que no hay suficiente personal de este tipo formado en el país, así como al envejecimiento de la población. Los gobiernos que deseen contener el éxodo de profesionales son libres de hacerlo, ya que esas medidas no están sujetas a las disciplinas del AGCS, las cuales se refieren -en particular en lo que respecta al modo 4 solo a la inmigración temporal de trabajadores sanitarios extranjeros. El alcance del modo 4 es limitado, tanto en su definición como en los compromisos concretos que establece, lo cual significa que, probablemente, el AGCS no tiene una repercusión significativa en la migración internacional de este tipo de personal.
4. Contratación pública
Se refiere en general a la adquisición de bienes, la contratación de servicios (incluso de construcción) o a la combinación de ambas por los organismos gubernamentales o en su nombre en cumplimiento de sus responsabilidades de servicio público, particularmente en áreas de enorme importancia social, como es el caso de la atención sanitaria. En esta sección se abordan los efectos positivos que un marco de contratación pública bien ideado puede tener sobre el sector sanitario. Se exponen asimismo las normas que el Acuerdo Plurilateral sobre Contratación Pública (ACP) de la OMC establece a tal efecto, así como la magnitud de los mercados de contratación en los sectores relacionados con la salud abarcados en el Acuerdo.101
Recuadro 2.13. Datos sobre la reducción de costos o la optimización de recursos en el sector de la atención sanitaria gracias a procesos de licitación transparentes y competitivos
|
Un estudio de 2011, publicado por la Oficina Nacional de Investigaciones Económicas de los Estados Unidos (Danzon et al., 2011) examinó los factores determinantes de los precios de los medicamentos originarios y genéricos en un número significativo de países. El estudio se centró principalmente en los medicamentos para el tratamiento de la infección por el VIH/sida, la tuberculosis y el paludismo en países de bajos ingresos y países de ingresos medianos. Se analizaron los efectos en los precios de los medicamentos cuando estos se venden al por menor en farmacias, por comparación con los casos en que se adquieren mediante licitaciones como las que realizan, por ejemplo, el Fondo Mundial y la Fundación Clinton. |
En el estudio se muestra que la contratación pública mediante licitación atrae a proveedores de medicamentos genéricos y reduce significativamente los precios de los medicamentos originarios y los genéricos con respecto a los precios de venta al por menor en farmacias. Concretamente, se concluye que: "Las pruebas relativas a los medicamentos contra la infección por el VIH/sida, la tuberculosis y el paludismo muestran que los mecanismos de contratación reducen los precios de los medicamentos originarios y los genéricos en un 42% y un 28%, respectivamente, en comparación con los precios al por menor en farmacias". |
En un estudio realizado por la OCDE en 2003 sobre los beneficios de los procesos de contratación transparentes y competitivos se mencionan los siguientes ejemplos de beneficios alcanzados:
|
|
(a) La importancia de los procesos de contratación transparentes y competitivos en el sector sanitario
La posibilidad de lograr ahorros significativos mediante la introducción de mejores herramientas de contratación o adquisición pública tiene una pertinencia especial en el sector sanitario, donde, según el Banco Mundial, la adquisición pública de medicamentos ha sido particularmente propensa a la mala administración, y ha contribuido al agotamiento de las existencias, al desperdicio, a la calidad deficiente y a la inflación de los precios (Banco Mundial, 2011). Análogamente, en un estudio sobre la fijación de precios de los medicamentos se concluyó que en las regiones de África, Europa y el Pacífico Occidental las administraciones pagaron entre un 34% y un 44% más de lo necesario, en promedio, por los medicamentos (Cameron et al., 2009). Ese tipo de deficiencias deben considerarse un fracaso notable de los sistemas de salud pública. Por el contrario, introducir procedimientos de adquisición pública más eficaces, transparentes y competitivos en esos sistemas puede mejorar sustancialmente la disponibilidad de medicamentos accesibles y asequibles, y ayudar a crear sistemas de prestación de atención sanitaria más eficaces y eficaces en función del costo, de manera que se reduzca el desperdicio y se eviten las prácticas fraudulentas y corruptas. En el recuadro 2.13 se presenta un resumen de datos sobre la reducción de costos conseguida mediante la aplicación de procesos de adquisición transparentes y competitivos en el sector de la atención sanitaria.
(b) Contratación de tecnologías médicas y servicios de salud con arreglo al Acuerdo sobre Contratación Pública
El Acuerdo sobre Contratación Pública (ACP) proporciona un marco apropiado para las normas internacionales destinadas a fomentar las buenas prácticas en esta esfera y el comercio eficaz. Como es un acuerdo plurilateral, solo los Miembros de la OMC que se han adherido han de atenerse a sus normas. En 2012 había 42 Miembros de la OMC que eran Partes en el ACP.
(i) Alcance del Acuerdo sobre Contratación Pública
El ACP tiene importantes aplicaciones en el sector público de atención sanitaria, y en concreto en los ámbitos que abarca, a saber, la adquisición de medicamentos y productos farmacéuticos y la contratación de servicios de salud. En principio, el ACP fomenta la transparencia y la competencia leal, y ayuda a los gobiernos y sus organismos a optimizar sus recursos. A menos que se establezca explícitamente alguna exclusión, se aplica a todos los bienes que las entidades abarcadas adquieren por valores superiores a los umbrales correspondientes102 , incluidos los medicamentos y los productos farmacéuticos (para conocer más detalles, véase el cuadro 2.5).
El ACP abarca únicamente los productos y servicios y los organismos o entidades gubernamentales que las Partes han designado específicamente e incluido en sus respectivas listas de compromisos en el Apéndice I del Acuerdo. Para determinar los compromisos en materia de acceso a los mercados que las Partes en el ACP han contraído específicamente en el sector de la atención sanitaria, deben tomarse en cuenta los siguientes factores: i) si la lista de compromisos de una Parte en el ACP abarca las entidades relacionadas con la salud, y, de ser así, qué entidades; y ii) si el ACP abarca los productos y servicios de salud, y, de ser así, qué productos y servicios.
Cuadro 2.5. Alcance del Acuerdo sobre Contratación Pública de la OMC en el sector sanitario para diversas Partes |
||||
|
Parte en el ACP de la OMC |
Alcance relativo a las entidades del gobierno central relacionadas con la salud |
Alcance relativo a las entidades del gobierno subcentral relacionadas con la salud |
Alcance relativo a los bienes (por lo general los productos farmacéuticos se consideran bienes) |
Alcance relativo a los servicios relacionados con la salud |
Armeniaa
|
P |
|
P |
P |
Canadá
|
P |
P |
P |
x |
Estados Unidos
|
P |
P |
P |
P |
Hong Kong,China |
P |
n.p. |
P |
x |
Islandiab
|
|
|
P |
x |
Israelc
|
P |
x |
P |
x |
Japón
|
P |
x |
P |
x |
Liechtenstein
|
|
|
P |
x |
Noruegaa
|
P |
|
P |
x |
Países Bajos, respecto de Aruba
|
P |
n.p. |
P |
x |
República de Corea
|
P |
x |
P |
x |
Singapur
|
P |
n.p. |
P |
x |
Suiza
|
P |
P |
P |
x |
Taipei Chino
|
P |
P |
P |
x |
Unión Europea, con inclusión de sus 27 Estados miembros
|
P |
P |
P |
x |
Observaciones: Los nombres de las Partes en el ACP son los que se utilizan en la OMC. Los símbolos "?" y "x" se han utilizado respectivamente para indicar si en el alcance correspondiente a una Parte se indica expresamente o no la inclusión de entidades relacionadas con la salud. Cuando el alcance de los compromisos de una Parte se ha formulado de manera genérica o descriptiva, sin proporcionar información más concreta -por ejemplo, mediante una lista ilustrativa- la entrada correspondiente se ha dejado en blanco. Se incluye asimismo una nota al pie para indicar que el elemento en cuestión no está expresamente incluido ni excluido. Cabe señalar que en los casos siguientes no existe gobierno subcentral y, en consecuencia, no se han consignado compromisos en ese nivel: Hong Kong, China; los Países Bajos respecto de Aruba; y Singapur. aEn el anexo 2, correspondiente a Noruega y a Armenia, no se incluyen ni se excluyen explícitamente las entidades relacionadas con la salud. bNo se hace mención expresa de la inclusión ni de la exclusión de las entidades relacionadas con la salud. cIsrael ha excluido expresamente los siguientes bienes adquiridos por el Ministerio de Salud: insulina y bombas de infusión, audiómetros, vendajes médicos (vendas, esparadrapo, excluidos vendas y apósitos de gasa), soluciones intravenosas, juegos para la administración de transfusiones, cánulas mariposa epicraneales, catéteres de hemodiálisis y sangre, bolsas de sangre y agujas hipodérmicas. Cabe señalar que varias de esas exclusiones se han suprimido debido a la conclusión de las negociaciones en el marco del ACP.
En relación con el primer aspecto, las Partes en el ACP incluyen entidades relacionadas con la salud a varios niveles de gobierno (véase el cuadro 2.5). Más concretamente:
- Casi todas las Partes incluyen expresamente las dependencias del gobierno central relacionadas con la salud (por ejemplo, entidades federales y ministerios).
- La mayoría de las Partes que tienen un nivel de gobierno subcentral (por ejemplo, estados, provincias, cantones o municipios) incluyen las entidades de ese nivel relacionadas con la salud, o no las excluyen expresamente.
- Tres de las Partes incluyen otro tipo de dependencias gubernamentales relacionadas con la salud (por ejemplo, hospitales).
Cabe señalar asimismo, tal como se aclara en el texto del ACP revisado, que el Acuerdo no se aplica a los bienes y servicios contratados con miras a la venta o reventa comercial.
Además, la Unión Europea ha contraído compromisos vinculantes en virtud del ACP para las dependencias relacionadas con la salud de los gobiernos centrales de sus 27 Estados miembros, así como para un número notable de dependencias relacionadas con la salud de los gobiernos subcentrales. Los Estados Unidos, por su parte, incluyen en el Acuerdo el Departamento de Salud y Servicios Sociales, de índole federal, así como entidades sanitarias de varios Estados.
Otro aspecto fundamental es que en el ACP los productos farmacéuticos se consideran generalmente bienes y, en consecuencia, a menos que se especifique lo contrario, se toman en cuenta cuando son adquiridos en cantidades superiores a los valores de umbral pertinentes por las entidades incluidas en las listas de las Partes. Además, ninguna de las Partes en el ACP incluye actualmente una exclusión general de los productos farmacéuticos en sus listas. Una Parte pequeña ha excluido ciertos bienes adquiridos por el Ministerio de Salud. En lo que respecta a la cobertura de los servicios relacionados con la salud en virtud del ACP, los Estados Unidos son actualmente la única Parte que los incluye. En resumen, el alcance del ACP es relativamente amplio en lo que respecta a las entidades del sector de la atención sanitaria, sobre todo con respecto a los bienes (en particular los medicamentos); por el contrario, en lo que respecta a los servicios de salud su alcance es limitado.
(ii) La magnitud de la contratación pública de las Partes en el ACP en la esfera de la salud
El ACP es el principal instrumento internacional que regula el comercio en los mercados de contratación pública; se estima que la magnitud total de la contratación pública realizada con arreglo al ACP fue de alrededor de 1,6 billones de dólares EE.UU. en 2008.103 A fin de valorar la importancia del alcance del ACP en los mercados de contratación pública relacionados con la salud, es necesario cuantificar la magnitud que pueden alcanzar esos compromisos en materia de acceso a los mercados. Actualmente existe una importante fuente de información estadística sobre la magnitud de los mercados de contratación pública, gracias a los últimos informes que las Partes en el ACP han presentado al Comité de Contratación Pública. A pesar de que esos informes no son necesariamente coherentes en todos los aspectos (se está tratando de lograr una mayor uniformidad en los criterios metodológicos), representan una fuente de información muy útil sobre la magnitud de los compromisos mencionados.104
Dichas fuentes oficiales dejan claro que la magnitud de los mercados de contratación pública en los sectores relacionados con la salud sujetos a la aplicación del ACP es considerable.105 Por ejemplo, los Estados Unidos indican en sus informes estadísticos que el gasto general total, por funciones, de los 37 Estados abarcados en el ACP fue en 2008 de 40.000 millones de dólares EE.UU. en hospitales y de 50.000 millones de dólares EE.UU. en salud.106 Asimismo, se calcula que ese año el valor de los bienes y servicios abarcados por el ACP y contratados por el Departamento de Salud y Servicios Sociales fue de alrededor de 30.000 millones de dólares EE.UU. Por su parte, la Unión Europea calcula en su informe estadístico de 2007 que las entidades abarcadas en el ACP adquirieron dispositivos médicos y de laboratorio, productos farmacéuticos e insumos médicos fungibles por valor de 11.000 millones de euros.107 Por último, el Japón señala que en 2010 el Ministerio de Salud, Trabajo y Bienestar adjudicó contratos en el marco del ACP por un valor estimado de 1.800 millones de dólares EE.UU.108
5. Acuerdos de libre comercio
(a) Tendencias actuales en las negociaciones comerciales más allá del ámbito multilateral
Los países de todo el mundo muestran una tendencia a establecer acuerdos de integración económica según diversas configuraciones bilaterales y regionales (véase el recuadro 2.14), en paralelo con su incorporación a acuerdos multilaterales, fenómeno que plantea importantes dificultades para el sistema multilateral que se describe en este capítulo (y se analiza en OMC, 2011). Esos acuerdos se conocen como acuerdos comerciales regionales, acuerdos de libre comercio, acuerdos comerciales bilaterales o (según el término utilizado por el Banco Mundial y la OMC en sus últimos informes) acuerdos comerciales preferenciales, ya que muchos de esos acuerdos no son "regionales" sino que pueden abarcar países geográficamente dispersos, y que prevén aranceles preferenciales sobre numerosos productos. Con frecuencia esos términos se solapan, y puede ocurrir que en la práctica varios de ellos se apliquen a un mismo acuerdo en función de las características de este. Para los fines del presente estudio, se utiliza el término "acuerdo de libre comercio" para referirse a todo tipo de acuerdo comercial.
En el pasado, los acuerdos de integración se centraban a menudo en el comercio de bienes y en la eliminación de aranceles y otras restricciones entre las partes. Sin embargo, la OMC (2011) señala que, en los últimos años, muchos acuerdos comerciales han adoptado la forma de procesos de integración profunda que incluyen disposiciones sobre una amplia gama de políticas internas o reglamentarias, tales como los servicios o la propiedad intelectual, así como una variedad mayor de agentes. La apertura del comercio que se deriva de esos procesos ejerce presión para que se concilien las prácticas nacionales divergentes, y crea exigencias en materia de gobernanza y de estado de derecho que trascienden las fronteras nacionales. En materia de derecho y política de propiedad intelectual, esa tendencia puede materializarse en importantes cambios en las leyes nacionales, que a su vez afectan directamente al marco por el que se rigen la innovación y el acceso a los medicamentos y a las tecnologías médicas. Todo ello conforma un conjunto de procesos que últimamente muestra más dinamismo que el establecimiento de normas en el ámbito multilateral.
Recuadro 2.14. La geografía que cambia y el alcance de los acuerdos de libre comercio |
Los acuerdos comerciales preferenciales pueden ser acuerdos de libre comercio o uniones aduaneras con aranceles exteriores comunes. Esta última "ola" de regionalismo abarca una red de participantes mucho más amplia -que incluye iniciativas bilaterales, plurilaterales e interregionales- y países con diferentes niveles de desarrollo económico, con inclusión de alianzas entre países desarrollados, entre países en desarrollo, y entre países desarrollados y países en desarrollo. Si bien esos nuevos acuerdos, al igual que los acuerdos comerciales preferenciales realizados con anterioridad, incluyen reducciones arancelarias preferenciales, se centran más en otras cuestiones, tales como las corrientes de capital, las normas, la propiedad intelectual, los sistemas reglamentarios (muchos de ellos no discriminatorios) y los compromisos sobre cuestiones laborales y ambientales. |
Intervienen en ello muchos factores; la OMC (2011) señala los siguientes: (i) neutralización de las políticas de "empobrecimiento del vecino", que buscan beneficiar a un país a costa de otros; (ii) incremento del tamaño del mercado; ( iii) aumento de la previsibilidad de las políticas; (iv) envío de una señal de apertura a los mercados; y (v) la expansión de las redes internacionales de producción. La OMC (2011) concluye que adoptar políticas comunes con las economías avanzadas puede ser beneficioso para los países en desarrollo, ya que les permite importar sistemas normativos que ya han sido "ensayados" y representan "prácticas óptimas". Por otra parte, los países en desarrollo pueden verse presionados para que adopten normas comunes no apropiadas para su nivel de desarrollo, o normas de las que las economías avanzadas podrían servirse para proteger sus intereses creados.
El aumento del tamaño del mercado puede ser una razón para establecer acuerdos comerciales preferenciales porque permite a las empresas de los Estados signatarios explotar las economías de escala y obtener una ventaja relativa sobre las empresas competidoras excluidas. Además, el acceso preferencial a mercados más grandes puede aumentar el atractivo de un país como destino de la inversión extranjera directa. Ambas razones tienen especial validez en el caso de economías pequeñas, lo que puede servir para explicar por qué esos países aceptan otorgar concesiones en otras cuestiones más polémicas, como los derechos de propiedad intelectual o las normas ambientales, al negociar acuerdos comerciales preferenciales con grandes economías (OMC, 2011).
(b) Bilateralismo y regionalismo: la cuestión de las preferencias
Una característica fundamental de los acuerdos de libre comercio es la idea del trato preferencial y de las ventajas para las partes en el acuerdo que pueden no ampliarse automáticamente a otras partes. En el caso de otras esferas que trascienden el ámbito de aplicación clásico de los acuerdos sobre el comercio de mercancías, tales como la contratación pública o la política de competencia, los negociadores también pueden estipular preferencias que benefician únicamente a las partes en el acuerdo. Sin embargo, la situación es diferente en lo que respecta a la mayoría de los aspectos de las normas sobre propiedad intelectual.
A diferencia de otros Acuerdos comerciales de la OMC, tales como el GATT o el AGCS, el Acuerdo sobre los ADPIC no prevé amplias excepciones al principio de la nación más favorecida en el caso de los acuerdos de libre comercio. Esto puede tener consecuencias importantes para el acceso a los medicamentos y a las tecnologías médicas, así como para la innovación de productos. En concreto, si dos Miembros de la OMC acuerdan conceder a sus respectivos nacionales un nivel de protección en materia de propiedad intelectual superior al estipulado en el Acuerdo, no pueden, en principio, denegar ese nivel de protección superior a los nacionales de los demás Miembros de la OMC. En otras palabras, el nivel superior de protección acordado no se limitaría a los nacionales de las partes en el acuerdo de libre comercio, sino que debería ampliarse igualmente a los nacionales de los demás Miembros de la OMC. Por ejemplo, si dos países acordaran conceder prórrogas del plazo de vigencia de las patentes a sus respectivos titulares de patente, el principio de la nación más favorecida con arreglo al Acuerdo exigiría que otorgaran las mismas prórrogas a los titulares de patente de los demás Miembros de la OMC. Por el contrario, si acordaran eliminar los aranceles sobre los productos farmacéuticos o los componentes químicos que se compran mutuamente, dentro de un acuerdo de libre comercio o unión aduanera, no tendrían que eliminar los aranceles sobre las importaciones de otros países.
(c) Normas de propiedad intelectual
Tal como se expuso en el apartado a) de la sección B.1 del capítulo II, y en la sección C.5 del capítulo IV, los Miembros de la OMC son libres de introducir en sus legislaciones nacionales una protección de la propiedad intelectual que trascienda las normas mínimas exigidas por el Acuerdo sobre los ADPIC, siempre y cuando esa protección no contravenga las prescripciones de este. Hay diversos acuerdos de libre comercio que prevén una protección mayor para las patentes y los datos de pruebas, así como normas de observancia más estrictas, que afectan al comercio de productos farmacéuticos y pueden repercutir sobre los precios de las tecnologías médicas. Muchos de esos acuerdos forman "familias", cada una de las cuales se agrupa en torno a un "centro". La Asociación Europea de Libre Comercio, los Estados Unidos y la Unión Europea son los "centros" más importantes desde el punto de vista de la cantidad de acuerdos que contienen disposiciones de ese tipo. Cada centro tiende a adoptar un criterio uniforme en la negociación de los acuerdos, por lo que las disposiciones en materia de propiedad intelectual (entre otras) de todos los acuerdos de cada familia a menudo comparten muchas características importantes. De hecho, de esta manera se exportan aspectos del régimen regulatorio del centro a sus socios comerciales. En ese sentido, la OMC (2011) señala que, en comparación con los Acuerdos de la OMC, ese proceso ha servido generalmente para elevar los niveles de compromiso. Las áreas que incorporan compromisos jurídicamente exigibles son relativamente pocas, y se encuentran principalmente en las esferas de las inversiones, la política de competencia, los derechos de propiedad intelectual y los movimientos de capitales.
Los acuerdos de libre comercio pueden llegar a tener amplias repercusiones en los regímenes nacionales de propiedad intelectual, ya que, como se ha indicado anteriormente, la mayor protección que exigen en esta materia, incluidas las patentes y los datos de pruebas, debe ponerse a disposición de los nacionales de los demás Miembros de la OMC sin discriminación, y no solo de los nacionales de la otra parte en el acuerdo. Por otro lado, en el caso de esferas que normalmente funcionan sobre la base de las reglamentaciones nacionales, como ocurre con la propiedad intelectual, de los servicios y de las políticas de competencia (OMC, 2011), en la práctica sería muy costoso adaptar la reglamentación a fin de favorecer a los nacionales de los socios preferenciales, dificultad que crece a medida que aumenta el número de acuerdos de libre comercio de los que un país es signatario. Por lo tanto, razones de principio y necesidades prácticas ocasionan un efecto de incremento gradual en las normas de propiedad intelectual, ya que se pueden congelar en los niveles más altos de protección, lo que puede repercutir en la innovación de las tecnologías médicas y el acceso a ellas. Se han publicado varias guías sobre acuerdos de libre comercio; por ejemplo, la Oficina Regional de la OMS para el Mediterráneo Oriental ha publicado una guía de política para negociadores y responsables de la aplicación de las disposiciones sobre propiedad intelectual en los acuerdos de libre comercio bilaterales (El Said, 2010).109
(d) Compromisos en otros sectores
Los acuerdos de libre comercio, por su propia naturaleza, no se limitan a establecer normas relativas a la protección y al ejercicio de los derechos de propiedad intelectual. Un análisis riguroso de los efectos que esos acuerdos pueden tener sobre la innovación y el acceso en materia de tecnologías médicas debe por lo tanto considerar las normas y los compromisos adoptados en otras esferas normativas fundamentales directamente relacionadas con el sector farmacéutico, como son las relativas a los aranceles, la contratación pública y la legislación sobre competencia.
Sin embargo, en lo que respecta a los aranceles, si bien los primeros acuerdos de libre comercio nacían con la intención de reducir, basándose en el trato de la nación más favorecida, unos aranceles relativamente elevados, es probable que en los últimos años la consecución de esas reducciones arancelarias, incluso para los productos farmacéuticos, haya perdido parte de la importancia que tenía en un principio, y que solo ocasionalmente sea considerada en los acuerdos de libre comercio. Según la OMC (2011), ello se debe a que, en 2009, el arancel medio aplicado entre distintos productos y países era solamente del 4%, de manera que no queda mucho margen para el intercambio de concesiones arancelarias preferenciales mediante acuerdos comerciales.
Por otro lado, cuestiones como las inversiones, la política de competencia y la contratación pública figuran cada vez con más frecuencia en los acuerdos de libre comercio más recientes, lo que complementa la reducción de los obstáculos al comercio y refleja una tendencia hacia la internacionalización de políticas que antes se trataban en el ámbito nacional. Las disciplinas conexas pueden abordarse bien sea en capítulos independientes del acuerdo, o bien, como suele suceder con la esfera de la competencia, pueden pasar a ser parte integral de determinados capítulos, por ejemplo, el relativo a los derechos de propiedad intelectual o a la contratación pública. La OMC (2011) estima, por ejemplo, que alrededor del 20% de los capítulos relativos a derechos de propiedad intelectual incorporan disposiciones para prevenir abusos de los derechos de propiedad intelectual o conductas anticompetitivas.
1. Para conocer un análisis de los aspectos económicos de la propiedad intelectual en la esfera de las tecnologías médicas, véase la sección C.5 del capítulo II. back to text
2. Se aborda más adelante, en la sección B.5. back to text
3. Ese efecto de "multilateralización" del alcance de los acuerdos bilaterales se aborda en la sección D del capítulo IV. back to text
4. Documento de la OMPI SCP/12/3 Rev.2. back to text
5. Ibid. back to text
6. Documento de la OMPI MTN.GNG/NG11/W/24/Rev.1. back to text
7. Para conocer una explicación más detallada de los criterios de patentabilidad, véase el subapartado iii) más adelante. back to text
8. Para consultar los pormenores del tratado, véase:www.wipo.int/treaties/en/registration/pct. back to text
9. Artículo 27 del PCT. back to text
10. Algunos acuerdos regionales destacados: el Convenio sobre la Patente Europea (CPE), la Convención Euroasiática sobre Patentes, el Protocolo de Harare de la Organización Regional Africana de la Propiedad Industrial (ARIPO), el Acuerdo de Bangui de la Organización Africana de la Propiedad Intelectual, la reglamentación en materia de patentes del Consejo de Cooperación del Golfo y la Decisión 486 del Régimen Común sobre Propiedad Industrial de la Comunidad Andina. back to text
11. El término "reivindicación" se explica en el subapartado vi) del apartado b) de la sección B.1. back to text
12. Véase el apartado b) de la sección C.1 del capítulo IV. back to text
13. La regla 29 del Reglamento de ejecución del Convenio sobre la Patente Europea, incluido en el CPE, aclara determinadas cuestiones en relación con la patentabilidad de las invenciones relativas al cuerpo humano, el uso de embriones humanos con fines industriales o comerciales y algunos otros casos en los que se excluye la concesión de patentes europeas.. back to text
14. Véase el documento G 002/06 (Use of embryos/WARF) de 25 de noviembre de 2008, OJ 2009, 306, en: www.epo.org/law-practice/case-law-appeals/pdf/g060002ep1.pdf. Véase asimismo el documento T 1374/04 (Stem cells/WARF) de 7 de abril de 2006, OJ 2007, 313, en: www.epo.org/law-practice/case-law-appeals/recent/t041374ex1.html. back to text
15. Véase el documento C-34/10, de 18 de octubre de 2011, en: http://curia.europa.eu/juris/liste.jsf?language=en&num=C-34/10. back to text
16. DO 1998 L213/13. back to text
17. Documento de la OMC IP/C/W/369/Rev.1. back to text
18. Esa cuestión se aborda con más detalle en el apartado a) de la sección D.3 del capítulo III. back to text
19. Esas cuestiones se abordan en el apartado b) de la sección D.3 del capítulo III. back to text
20. Ibid. back to text
21. Ibid. back to text
22. Documento de la OMPI SCP/13/5. back to text
23. Véase el ejemplo de la decisión del Tribunal Supremo del Canadá de 8 de noviembre de 2012, 2012 SCC, 60, Teva Canada Ltd. contra Pfizer Canada Inc., en: http://scc.lexum.org/decisiascc-csc/scc-csc/scc-csc/en/item/12679/index.do. El Tribunal, en su decisión del 8 de noviembre de 2012 [EN 70] sostuvo que la patente del Canadá 2,163,446, concedida por una invención para el tratamiento de la impotencia, era nula porque la solicitud de patente no cumplía con las prescripciones de divulgación establecidas en la Ley de Patentes del Canadá (R.S.C. 1985, c. P-4). El Tribunal declaró que una adecuada divulgación en la descripción de la patente era una condición previa para concederla. La descripción, que incluía las reivindicaciones y la divulgación, debía concretar el alcance preciso y exacto del derecho que reivindicaba. El lector, desde la perspectiva de una persona del oficio, y basándose solamente en la descripción de la patente, debía estar en situación de hacer la misma utilización que el inventor podía hacer en el momento en que solicitó la patente. En el caso que se describe, las reivindicaciones estaban estructuradas "en cascada": la reivindicación Nº 1 se refería a más de 260 trillones de compuestos, las reivindicaciones 2 a 5 abarcaban una cantidad de compuestos cada vez menor, y las reivindicaciones 6 y 7 se referían cada una a un único compuesto. El Tribunal declaró que la práctica de las reivindicaciones en cascada era común y no interfería necesariamente con la prescripción de divulgación. El lector versado sabía que cuando una patente contiene reivindicaciones en cascada, la reivindicación pertinente es por lo general la última, referida a un único compuesto. Los compuestos que no funcionaban se consideraban no válidos, simplemente, y las reivindicaciones válidas seguían adelante. Sin embargo, en el caso descrito las dos últimas reivindicaciones hacían referencia a un compuesto cada una, de manera que una persona del oficio no podía determinar, basándose solamente en la información divulgada en la descripción de la patente, si era la reivindicación Nº 6 o la Nº 7 la que hacía referencia al compuesto eficaz. Hacían falta más pruebas para determinar cuál de esos dos compuestos era el realmente eficaz. Así pues, la información proporcionada en la patente no exponía con claridad en qué consistía la invención, sino que la información se presentaba de forma oscura. back to text
24. El "estado de la técnica" es, en general, todo conocimiento puesto a disposición del público antes de la presentación de la solicitud o la fecha de prioridad de una solicitud de patente en examen. Se utiliza para determinar el alcance de la novedad y de la actividad inventiva, dos requisitos de patentabilidad (documento de la OMPI SCP/12/3 Rev.2). back to text
25. Documentos de la OMPI SCP/12/3 y CDIP/7/3. back to text
26. Los procedimientos de concesión de patentes, desde la perspectiva del acceso a los medicamentos, se abordan en las secciones C.1 y C.2 del capítulo IV. back to text
27. Para obtener más información sobre sistemas de oposición y otros mecanismos administrativos de revocación o invalidación, véase el documento de la OMPI SCP/18/4. Los procedimientos de examen, desde la perspectiva del acceso a los medicamentos, se abordan en la sección C.2 del capítulo IV. back to text
28. La cuestión de las licencias se aborda con más detalle en el apartado c) de la sección D.4 del capítulo III, y en el apartado d) de la sección C.3 del capítulo IV. back to text
29. Para obtener más información, véase el subapartado vi) del apartado a) de la sección C.5 del capítulo IV. back to text
30. Véanse asimismo los documentos de la OMPI SCP/13/3, SCP/15/3, SCP/16/3, SCP/17/3 y SCP/18/3. Las excepciones y limitaciones y las flexibilidades previstas en el sistema de patentes, desde la perspectiva de la innovación y el acceso a los medicamentos, se abordan en la sección D.4 del capítulo III y en el apartado a) de la sección C.3 del capítulo IV, respectivamente. back to text
31. Véase el apartado b) de la sección D.4 del capítulo III. back to text
32. Las licencias obligatorias se abordan en los subapartados i) y ii) de la sección C.3 del capítulo IV. back to text
33. La información sobre patentes, desde la perspectiva de la innovación y el acceso a los medicamentos, se aborda en el apartado f) de la sección D.4 del capítulo III y en la sección C.4 del capítulo IV. back to text
34. Véase la publicación en línea Manual de la OMPI de información y documentación en materia de Propiedad Industrial:, www.wipo.int/standards/en/. back to text
35. Para consultar una lista de normas, recomendaciones y directrices de la OMPI, véase: www.wipo.int/standards/en/part_03_standards.html. back to text
36. Para obtener más información, véanse el Manual de la OMPI de información y documentación en materia de propiedad industrial (en: www.wipo.int/standards/en/pdf/08-01-01.pdf);y las definiciones de familia de patentes de la Oficina Europea de Patentes (en: www.epo.org/searching/essentials/patent-families.html). back to text
37. Se proporciona una visión general de las cuestiones relativas a la libertad para operar en el apartado g) de la sección D.4 del capítulo III. back to text
38. En un estudio técnico de la OMPI (documento CDIP/4/3 REV./STUDY/INF/3) se analizó la disponibilidad de los datos sobre la situación jurídica tanto en fuentes primarias como en fuentes secundarias, y se expusieron las dificultades asociadas a la disponibilidad, fiabilidad y comparabilidad de los datos. Aportaron datos al estudio un total de 87 autoridades de patentes, quienes confirmaron la situación, en ocasiones deficiente, del acceso a datos fiables sobre la situación jurídica de las patentes y de la comparabilidad de dichos datos. El estudio incluye recomendaciones de mejora, que exigirían un esfuerzo considerable por parte de las autoridades nacionales. Se puede obtener más información acerca del Proyecto de la OMPI sobre la situación jurídica de las patentes en la dirección www.wipo.int/patentscope/en/programs/legal_status/index.html. back to text
39. El título completo del libro es Approved Drug Products with Therapeutic Equivalence Evaluations, y se puede consultar en: www.fda.gov/Drugs/InformationOnDrugs/ucm129662.htm. back to text
40. Véase www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=314.53. back to text
41. Véase www.hc-sc.gc.ca/dhp-mps/prodpharma/patregbrev/index-eng.php. back to text
42. Fuente: Medicines Patent Pool, en: www.medicinespatentpool.org/patent-data/. back to text
43. Véanse la sección B.5 del capítulo II y la sección C.5 del capítulo IV. back to text
44. Se explican en el subapartado iii) de la sección C.3 del capítulo IV. back to text
45. Ibid. back to text
46. Directiva 2004/27/CE del Parlamento Europeo y del Consejo Europeo, de 31 de marzo de 2004, que modifica la directiva 2001/83/CE, por la que se establece un código comunitario sobre medicamentos de uso humano. back to text
47. Véase http://www.wipo.int/madrid/en/. back to text
48. Véase www.who.int/medicines/publications/druginformation/innlists/en/index.html. In addition, the INN Extranet, MedNet, grants free access to the INN searchable database: http://mednet.who.int/. back to text
49. Documento de la OMPI SCP/19/4. back to text back to text
50. Otros países, como Australia, el Canadá, el Japón, México y Sudáfrica, han creado, en sus ministerios de salud, sus propios procedimientos de examen de las marcas registradas. back to text
51. El Servicio de la FDA de prevención y análisis de errores de medicación (DMEPA) y el Grupo de Revisión de las Denominaciones de Fantasía (NRG). back to text
52. El NRG rechazó el 47% de los nombres propuestos en 2011. Véase el comunicado de prensa EMA/802336/2011 de la Agencia Europea de Medicamentos, de 2011: Overview of Invented Names Reviewed in November 2011 by the Name Review Group (NRG). El DMEPA rechaza aproximadamente un tercio de las marcas registradas propuestas (Scheib y Witherell, 2011). back to text
53. Caso Nº 594/2000: Sentencia del Tribunal Supremo de Pretoria, de 25 de marzo de 2002, favorable a Beecham Group plc y SmithKline Beecham Pharmaceuticals (Pty) Ltd, demandantes, contra Biotech Laboratories (Pty) Ltd, demandado. El Tribunal consideró que los demandantes habían demostrado que el prospecto podía calificarse de obra literaria según la definición de la Ley de Derechos de Autor de Sudáfrica, y prohibió a Biotech infringir esos derechos. Biotech Laboratories (Pty) Ltd apeló la decisión del Tribunal, que desestimó el recurso, con costas. back to text
54. Caso Nº FCA 1307: Sentencia del Tribunal Federal de Australia, de 18 de noviembre de 2011, por la que se establece que no procede reparación alguna por infracción de derechos de autor para los demandantes: Sanofi-Aventis Australia Pty Ltd, Sanofi-Aventis Deutschland Gmbh y Aventisub II Inc., contra el demandado Apotex Pty Ltd. back to text
55. Véase el recuadro 4.10 del capítulo IV. back to text
56. Ibid. back to text
57. Véanse los documentos de la OMC IP/C/W/570 e IP/C/W/571; y European Commission (2010). back to text
58. Véase el apartado e) de la sección C.3 del capítulo IV. back to text
59. Los avances en relación con la aplicación de la protección de la propiedad intelectual y su vinculación con el acceso a las tecnologías médicas, y en particular la repercusión de las normas de observancia más estrictas, derivadas de marcos normativos nacionales o interregionales tales como el Acuerdo Comercial de Lucha contra la Falsificación (ACTA), se abordan en el capítulo IV. back to text
60. Documento de la OMPI CDIP/5/4 Rev. back to text
61. Documento de la OMC WT/L/540. Véanse el subapartado iii) de la sección C.3 del capítulo IV y el anexo II. back to text
62. Véase www.wipo.int/treaties/en/agreement/trtdocs_wo030.html. back to text
63. Documento de la OMPI CDIP/5/4 Rev. back to text
64. Ibid. back to text
65. Documento de la OMPI CDIP/7/3 and CDIP/7/3 Add. back to text
66. Véase www.wipo.int/treaties/en/agreement/trtdocs_wo030.html. back to text
67. Véase Chapter III, Section D.4(b). back to text
68. Véase Chapter III, Box 3.8. back to text
69. Véase Chapter IV, Section C.3(iii). back to text
70. Véase Chapter II, Section B.1(g)(v). back to text
71. Véase Chapter IV, Section C.3(a)(i). back to text
72. El informe se puede consultar en:http://whqlibdoc.who.int/hq/2001/a73725.pdf. back to text
73. Se puede consultar el acta de la sesión extraordinaria en el documento de la OMC IP/C/M/31. back to text
74. Esta cuestión se explica en el apartado c) -de la sección C.3 del capítulo IV. back to text
75. Véase el subapartado iii) del apartado a) de la sección C.3 del capítulo IV. back to text
76. Documentos de las Naciones Unidas A/RES/65/1 y A/RES/65/277. back to text
77. Documento de la OMC IP/C/25. back to text
78. Documento de la OMC WT/L/478. back to text
79. Documento de la OMC IP/C/40. back to text
80. Documento de la OMC WT/L/845. back to text
81. Documento de la OMC IP/C/W/583. back to text
82. Párrafo 8 del artículo 18 de la Ley Nº 31/2009, de 26 de octubre de 2009, sobre la protección de la propiedad intelectual. back to text
83. Véanse el subapartado v) del apartado g) de la sección B.1 del capítulo II y la sección B.6 del capítulo IV. back to text
84. Véase www.wipo.int/wipolex/en/other_treaties/details.jsp?treaty_id=227. back to text
85. Documento de la OMC LT/UR/A/2. back to text
86. Documento de la OMC WT/ACC/UKR/152, paras. 425, 433 and 512. back to text
87. Documento de la OMC WT/L/508. back to text
88. Documento de la OMC WT/L/846. Véase www.wipo.int/wipolex/en/other_treaties/details.jsp?treaty_id=227. back to text
89. Documento de la OMC WT/L/508/Add.1. back to text
90. Véase: World Bank (2005, 2009). Para conocer una lista de publicaciones de la OCDE, véase:www.oecd.org/regreform/liberalisationandcompetitioninterventioninregulatedsectors/bestpracticeroundtablesoncompetitionpolicy.htm. En concreto, véanse los documentos correspondientes a las siguientes mesas redondas de políticas: Generic Pharmaceuticals (2009); Competition, Patents and Innovation II (2009); Competition, Patents and Innovation (2006); Competition in the Provision of Hospital Services (2005); Enhancing Beneficial Competition in the Health Professions (2004); Competition in the Pharmaceutical Industry (2000); y, desde una perspectiva más general, Relations Between Regulators and Competition Authorities (1998). back to text
91. Se ha señalado que las fusiones recientes entre empresas farmacéuticas han dado lugar a una reducción de las actividades de investigación y desarrollo en el sector. Véase, por ejemplo, LaMattina (2011). back to text
92. El término "condiciones exclusivas de retrocesión" se refiere a toda obligación del licenciatario de conceder al licenciante una licencia exclusiva sobre las mejoras que él mismo realice sobre la tecnología objeto de la licencia, o sobre las nuevas aplicaciones que haya obtenido a partir de dicha tecnología. Las "condiciones que impidan la impugnación de la validez" son aquellas que imponen al licenciatario la obligación de no impugnar la validez de los derechos de propiedad intelectual que sean titularidad del licenciante. Las "licencias conjuntas obligatorias" se refieren a la obligación del licenciatario de aceptar una licencia sobre varias tecnologías diferentes aunque solo esté interesado en una parte de esas tecnologías. back to text
93. A este respecto, varios organismos nacionales o regionales encargados de la competencia han publicado directrices que proporcionan una base sólida para el análisis de los casos en materia de propiedad intelectual y defensa de la competencia: US Department of Justice/Federal Trade Commission (1995); Comisión Europea, Directrices relativas a la aplicación del artículo 81 del Tratado CE a los acuerdos de transferencia, documento CE 2004/C 101/02; para el Canadá, véase Competition Bureau (2000); se puede consultar una traducción oficiosa del documento Guidelines for the Use of Intellectual Property under the Antimonopoly Act de la Comisión de Comercio Leal del Japón en la dirección:www.jftc.go.jp/en/legislation_guidelines/ama/pdf/070928_IP_Guideline.pdf, el documento Guidelines on Standardization and Patent Pool Arrangements se puede consultar en: is available at www.jftc.go.jp/en/legislation_guidelines/ama/pdf/Patent_Pool.pdf y el documento Guidelines Concerning Joint Research and Development under the Antimonopoly Act en:www.jftc.go.jp/en/legislation_guidelines/ama/pdf/jointresearch.pdf. back to text
94. Para obtener más información acerca de los datos sobre aranceles, véase la sección D.1 del capítulo IV. back to text
95. Véase www.haiweb.org/medicineprices. back to text
96. Para obtener más información, véase OMS/OMC (2002). back to text
97. Ibid. back to text
98. Esas descripciones por sectores se pueden encontrar en la Lista de clasificación sectorial de los servicios (documento de la OMC MTN.GNS/W/120), que los Miembros de la OMC han utilizado de forma general para consignar sus compromisos en el marco del AGCS. Los sectores i) a iii) indicados más arriba se incluyen en la sección relativa a "Servicios sociales y de salud"; los sectores iv) y v), en "Servicios profesionales", y el vi) en "Servicios financieros". back to text
99. Si se tienen en cuenta las limitaciones horizontales consignadas en algunas listas (es decir, limitaciones que abarcan todos los sectores incluidos), predominan los compromisos parciales. back to text
100. OMS, Migration of Health Workers, Hoja descriptiva Nº 301, 2010. back to text
101. Para obtener más información, véase: Müller y Pelletier (en preparación). back to text
102. El contenido completo de las listas de las Partes en el Acuerdo sobre Contratación Pública (apéndice 1), incluidos los umbrales pertinentes, se puede consultar en: www.wto.org/english/tratop_e/gproc_e/gp_gpa_e.htm. back to text
103. Véase http://shenyang.usembassy-china.org.cn/wto-gpa.html. back to text
104. Se pueden consultar más datos estadísticos en:www.wto.org/english/tratop_e/gproc_e/gp_gpa_e.htm. back to text
105. Cabe señalar que el siguiente análisis se centra únicamente en las oportunidades de acceso al mercado previstas en el marco del Acuerdo sobre Contratación Pública. No toma en cuenta los obstáculos para el acceso a los mercados que pudieran surgir de ámbitos ajenos al Acuerdo (por ejemplo, del ámbito de los derechos de propiedad intelectual). back to text
106. Documento de la OMC GPA/102/Add.3. Conviene recordar que el Acuerdo sobre Contratación Pública abarca las entidades, bienes y servicios especificados en las listas de compromisos de cada una de las Partes. back to text
107. Documento de la OMC GPA/94/Add.4. back to text
108. Documento de la OMC GPA/108/Add.4. El valor notificado se expresó en derechos especiales de giro y se ha convertido a dólares de los Estados Unidos. El valor estimado puede verse afectado por las variaciones en los tipos de cambio y problemas de conversión conexos. back to text
109. Algunas de las características particulares de los acuerdos de libre comercio pertinentes para los productos farmacéuticos se abordan en la sección C.5 del capítulo IV. back to text